1. Introduction
The purpose of this vignette is to guide users through the basic capabilities of the ‘divDyn’ package. Fossil occurrence databases, such as the Paleobiology Database (PaleoDB, https://paleobiodb.org/) are readily available to be used in analyses of diversity, extinction and origination patterns (the dynamics of biodiversity), with a certain toolkit that has become standard since the creation of the database. Until now, the implementation of most of these tools have been the responsibilities of individual researchers, with no software package to rely on. This R package intends to fill this gap.
1.1. Installation
To install this beta version of the package, you must download it either from the CRAN servers or its dedicated GitHub repository (https://github.com/divDyn/r-package/). All minor updates will be posted on GitHub as soon as they are finished, so please check this regularly. The version on CRAN will be lagging for some time, as it takes the servers many days to process everything and updates are expected to be frequent. All questions should be addressed to Adam Kocsis, the creator and maintainer of the package (adam.kocsis@fau.de). Instead of spending it on actual research, a tremendous amount of time was invested in making this piece of software useable and user-friendly. If you use a method implemented in the package in a publication, please cite both its reference(s) and the ‘divDyn’ package itself (Kocsis et al. 2019).
2. Necessary Data
Most functionality in the ‘divDyn’ package assumes that the time dimension is broken down to discrete intervals. Accordingly, most functions are built on two fundamental data structures: a time scale table and an occurrence dataset.
2.1. Time scales
The workflow presented here is based on the discretization of geological time, which is constrained by stratigraphy. These intervals of time (bins) represent the basic units of the analysis, and their sequence is coded in the time scale table. Even if we develop a geological model that outputs robust estimates in a continuous time axis, the calculation of metrics presented in the package will require discretization. We added implementations of the basic functionalities for continuous time (chapter ‘4.3. Slicing’) as well, but we do not deem it as reliable as using stratigraphic bins for million-year-scale, deep-time analyses. As age estimates are dependent on the different geological ‘time scales’, binning the data can change more than necessary, which can have random effects on the resulting series.
In order to demonstrate the workflow of binned analyses, we added an
example table to the package. This table represents a somewhat altered
form (see below) of the stage-level geological time scale of Gradstein
et al. (2020). You can attach this table using the data()
function.
library(divDyn)
# attach the time scale object
data(stages)Every row in this table represents a bin in the timescale. The most
important variable in this table is the slice number (in this case
num). This variable links every occurrence to one of the
bins. You can gather additional information by typing
?stages to the console. You can visualize the timescale by
using the plots() function included in the package:
tsplot(stages, boxes="sys", shading= "series")
For easier navigation in the plot, the time dimension can be
indicated with three variables: the radiometric dates that serve as
coordinates; boxes of intervals under lowest ylim value of
the plot; and vertical shades over the plotting area. The time scale to
be plotted can be altered by changing the values of the main argument
tsdat and by providing the appropriate column names for the
boxes and shading arguments. In order to use the system (period) names
as labels and the stages as shades, just change the function input
accordingly (the xlim values will limit the x axis
plot):
tsplot(stages, boxes="system", shading="stage", xlim=59:81)
The function was designed to enable the highest level of
customization. You can customize the distribution of plotting area with
the xlim (accepts exact ages and sequences of bins, see the
examples), ylim, prop and gap
arguments, and the color of the shading. You can also customize the
characteristics of the general plotting (calling plot(),
the boxes of time slices (calling rect()) and the labels
within them (calling the text() function). You can directly
control the arguments of these functions that tsplot() uses
to draw the elements of the timescale by adding the additional arguments
as lists to the plot.args, boxes.args and
labels.args arguments.
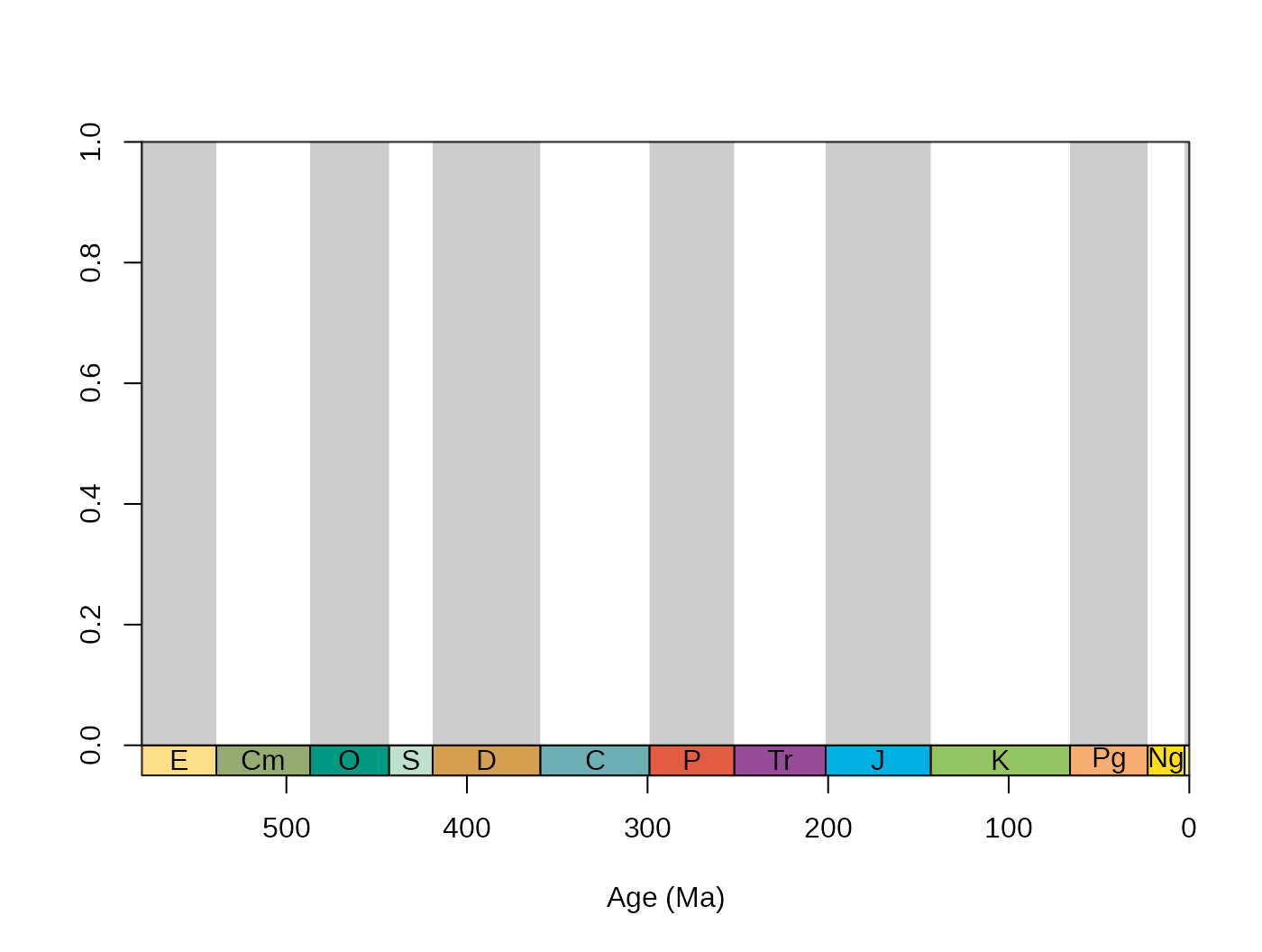
For instance, if you want your boxes to feature red italic fonts as
labels, just add col=“red”, and font=3 the way you would regularly use
them with the text() function, but wrap them up in a list, and assign it
to the ‘labels.args’ argument:
tsplot(stages, boxes="sys", shading="series",
labels.args=list(col="red", font=3), shading.col=c("white", "wheat"))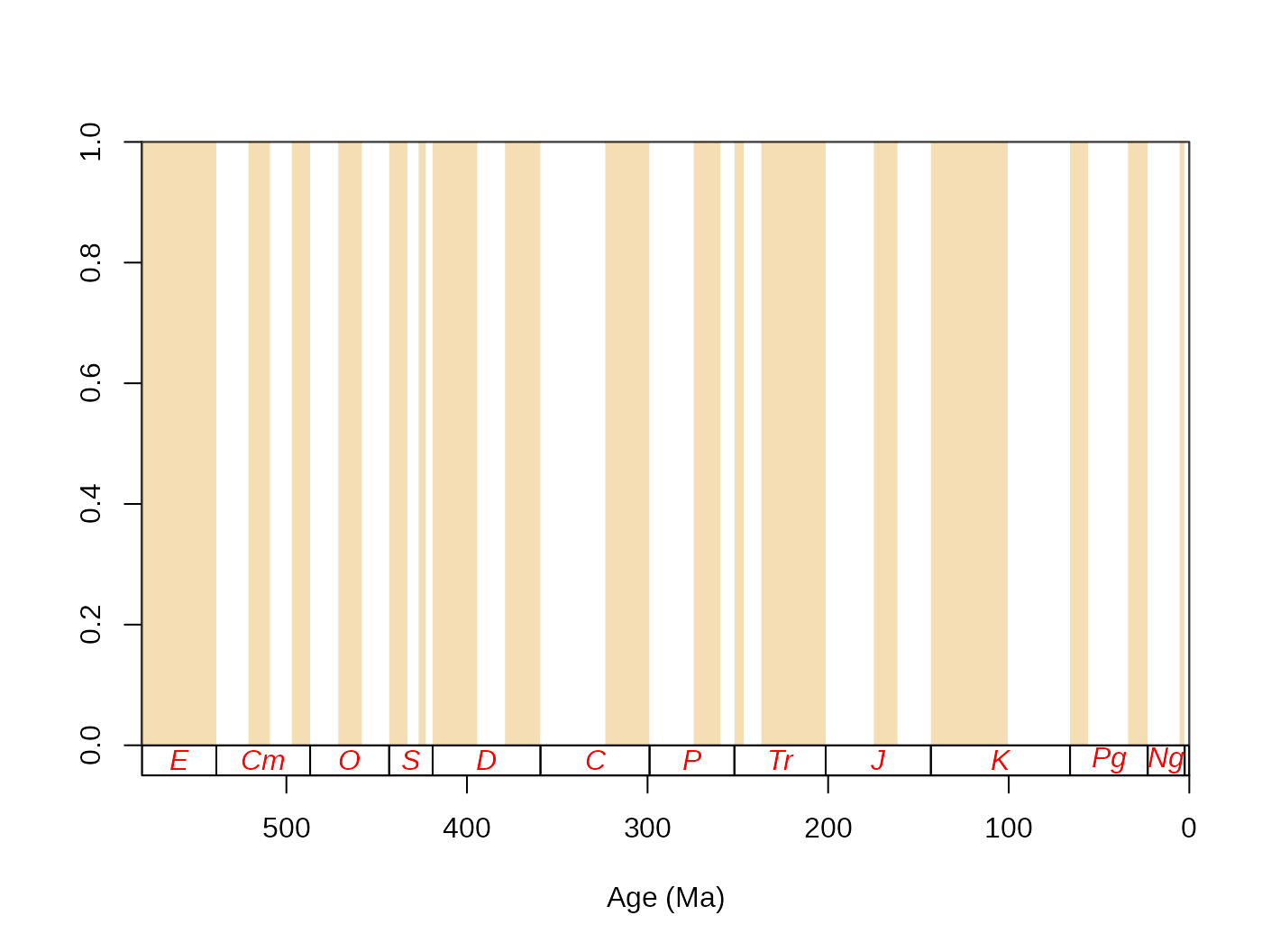
Although you can set it manually with the boxes.args
argument, the time scale plotting function also features colored
plotting of the boxes, via the argument boxes.col. The
stages object contains hexadecimal RGB codes for the system
(period), series (epoch) and stage (age) entries of the ICS table of
stratigraphy in the systemCol, seriesCol and
col columns, respectively.
Plotting of multiple levels as boxes is also supported now. Use vectors of column names if you want to add these.
tsplot(stages, boxes=c("short","system"), shading="short",
xlim=59:69, boxes.col=c("col","systemCol"), labels.args=list(cex=0.5))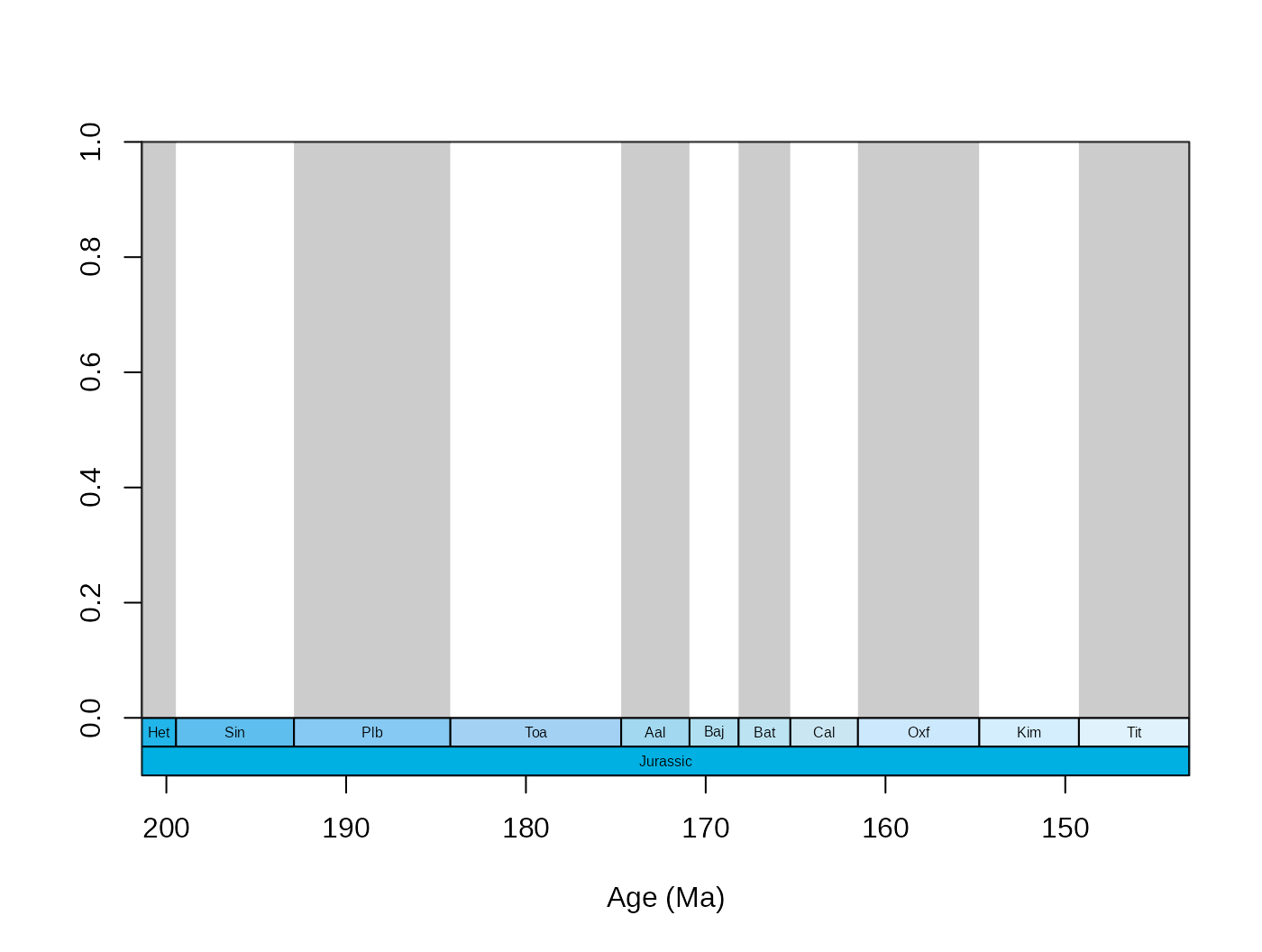 Accordingly, the
Accordingly, the boxes.args and labels.args
accept hierarchical input that corresponds to the levels of
stratigraphic hierarchy.
tsplot(stages, boxes=c("short","system"), shading="short",
xlim=59:69, boxes.col=c("col","systemCol"),
labels.args=list(list(cex=0.5),list(cex=1)),
boxes.args=list(list(),list(density=80)))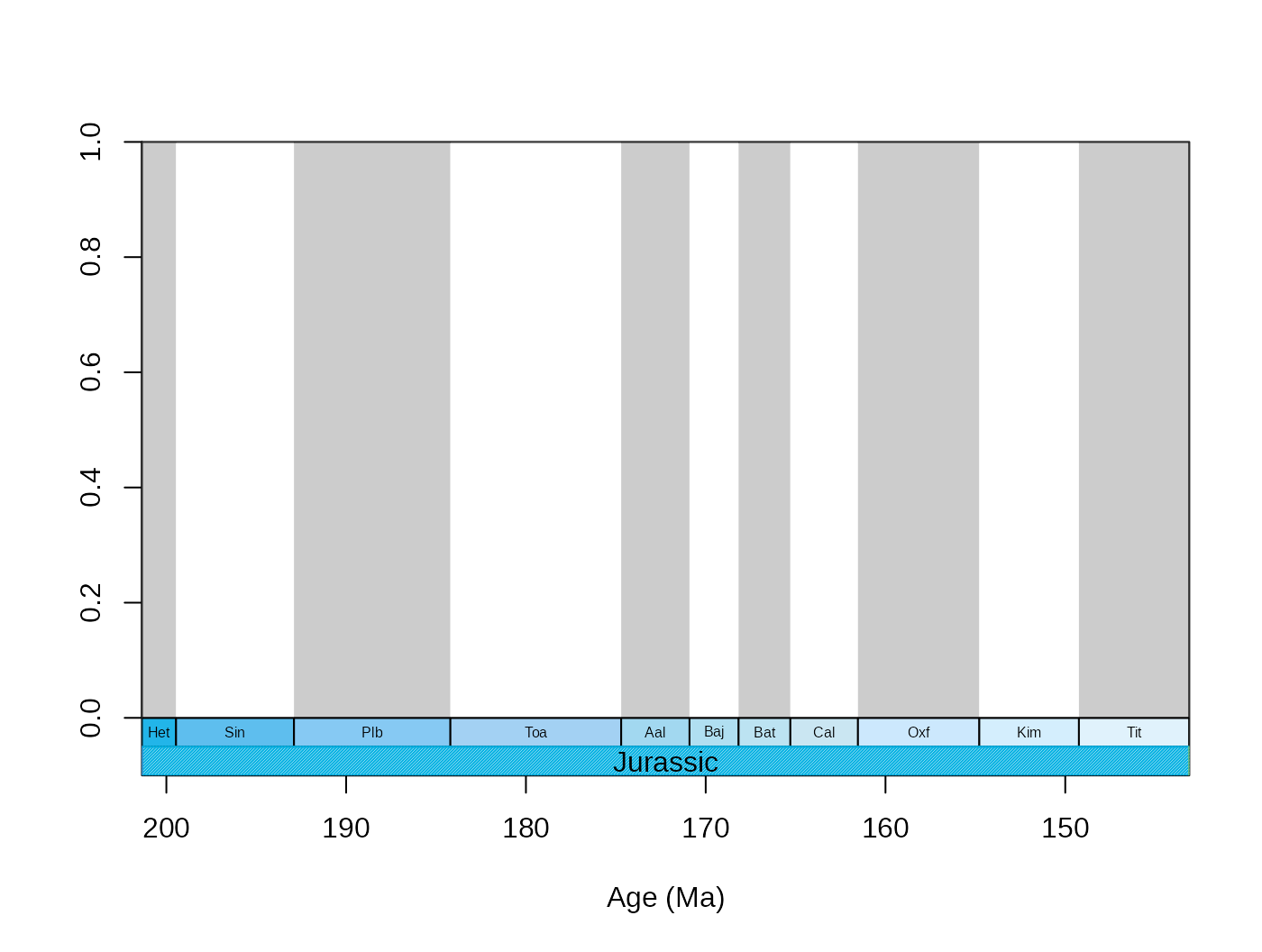 Naturally, you can use other time scale plotting packages such as the
‘geoscale’ package developed by J. Bell (2015). These work great for
Phanerozoic scale analyses, but be sure to check the compatibility of
the time scale you use for the binning and the time scale used for
plotting. As we have experience with problems that stemmed from the
incompatibility of analyzing and plotting data, we recommend using your
own timescale for plotting.
Naturally, you can use other time scale plotting packages such as the
‘geoscale’ package developed by J. Bell (2015). These work great for
Phanerozoic scale analyses, but be sure to check the compatibility of
the time scale you use for the binning and the time scale used for
plotting. As we have experience with problems that stemmed from the
incompatibility of analyzing and plotting data, we recommend using your
own timescale for plotting.
2.2. Occurrence table
The occurrence tables contain unary information about the presence of a taxon at a specified locality (can be global). In these tables, each occurrence is represented by a row which has to include a name of the taxon. This data format is similar to the following example:
## tax bin locality
## 1 Sp1 1 first
## 2 Sp1 1 second
## 3 Sp1 2 second
## 4 Sp2 2 second
## 5 Sp3 2 first
## 6 Sp2 3 secondThe functions of the package will have to be pointed to this column,
by specifying its name in the tax argument of the function
in question. Additional variables can be added that contain specific
information about the time and locality of the occurrence, as well as
other variables that help with grouping the individual entries
(taxon/collection information). The utility of this long format is in
its unbounded nature, with the acquisition of newer data points the time
and spatial coverage of the dataset can extend without problems.
2.2.1. Stratigraphic assignment
Most functions rely on processes that subset the data to contain
occurrences that represent the same time interval. This column can be
specified with setting the bin argument accordingly.
However, to get this column, a number of processes has to be run on the
raw data. Although the package already incorporates functions and data
to assign downloaded occurrences to stratigraphic bins, those are
illustrated in a separate vignette (https://github.com/divDyn/ddPhanero).
2.2.2. The example file
In order to demonstrate most capabilities of the package we have
added a fossil occurrence table of Scleractinian corals that we used in
an earlier study (Kiessling and Kocsis, 2015). This subset was
downloaded from the PaleoDB and was extended with information on
inferred photosymbiotic status, growth types, degree of integration,
ecological environment, inferred depth, substrate lithology and
latitudinal groups. Additional details are available by typing
?corals to the console. This dataset is embedded in the
package and can be attached using the data() function:
data(corals)This dataset was resolved to the 10 my timescale of the PaleoDB
(ten, now only available through FossilWorks) and the
stage-level time scale (variable stg) that is included in
the package (see Section 2.1, stages). This latter is the
basis of all inference and plotting. The values of the stg
column of the coral table refers to entries in the num
column of the stages table. Please note that this dataset
does not include Holocene occurrences. The occurrences designated with
stg==95 are just single entries that include extant genera;
therefore all other entries of this subset are missing, except for the
variables linked to the taxa. The rest of the occurrences represent
actual fossils.
fossils <- corals[corals$stg!=95,]
# the number of occurrences
nrow(fossils)## [1] 295443. Basic patterns
3.1. Ranges
We can gain preliminary knowledge by examining the basic patterns of the stratigraphic ranges . Probably the most apparent of these are the stratigraphic ranges of the taxa, which can be easily summarized in the FAD-LAD matrix:
fl <- fadlad(fossils, bin="stg", tax="genus")You can also use the occurrences’ own age estimate to calculate the
ranges, just type in ?fadlad to see some more examples. The ranges of
fossil taxa are the primary data quality feedback we can have from the
massive amount of fossil occurrences. You can easilly visualize these
with the ranges() function. To keep things simple, just
assign the age (stage) mean age to the occurrences:
fossils$mid <- stages$mid[fossils$stg]
tsplot(stages, shading="series", boxes="sys",xlim=c(260,0))
ranges(fossils, tax="genus", bin="mid")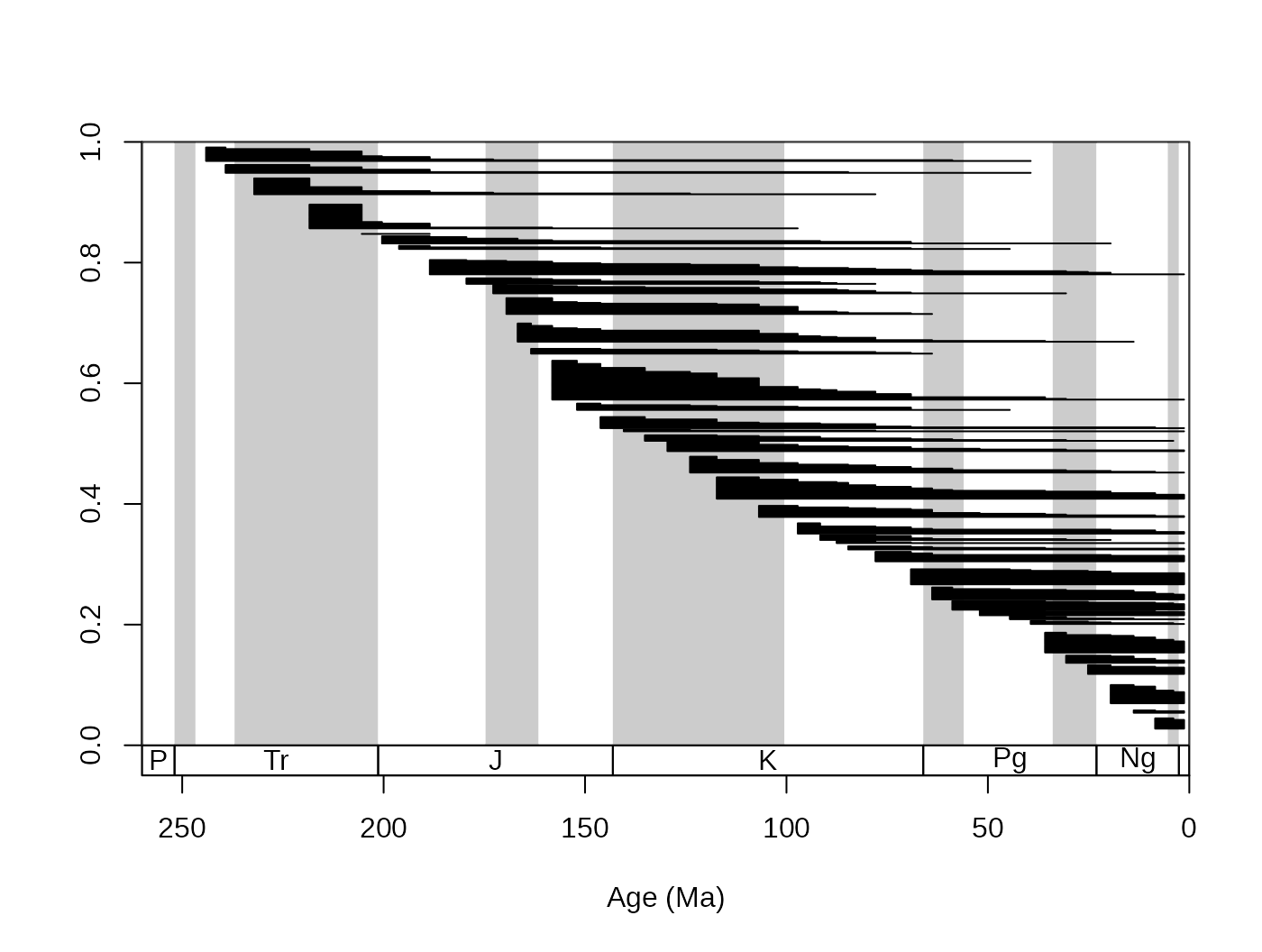
The function automatically selects the taxa that have ranges that
fall into the xlim values of the open device (you can
suppress this if you want to). If you zoom in with the main plotting
function, you can see this effect. You can also add the taxon labels by
setting the labs argument to TRUE.
tsplot(stages, shading="series", boxes="series",xlim=c(100,81))
ranges(fossils, tax="genus", bin="mid", labs=T, labels.args=list(cex=0.2))
As you can see from the example above (labs.args
argument), the argumentation of this function works in a similar way to
the that of the tsplot() function. You can get an even more
detailed look if you set the filt argument to
"orig". This will limit the displayed taxa to those that
originated within the interval. occs=TRUE will also plot
the sampled occurrences on the ranges.
tsplot(stages, shading="stage", boxes=c("stage","series"),xlim=c(100,80))
ranges(fossils, tax="genus", bin="mid", labs=T,
labels.args=list(cex=0.6), filt="orig", occs=T)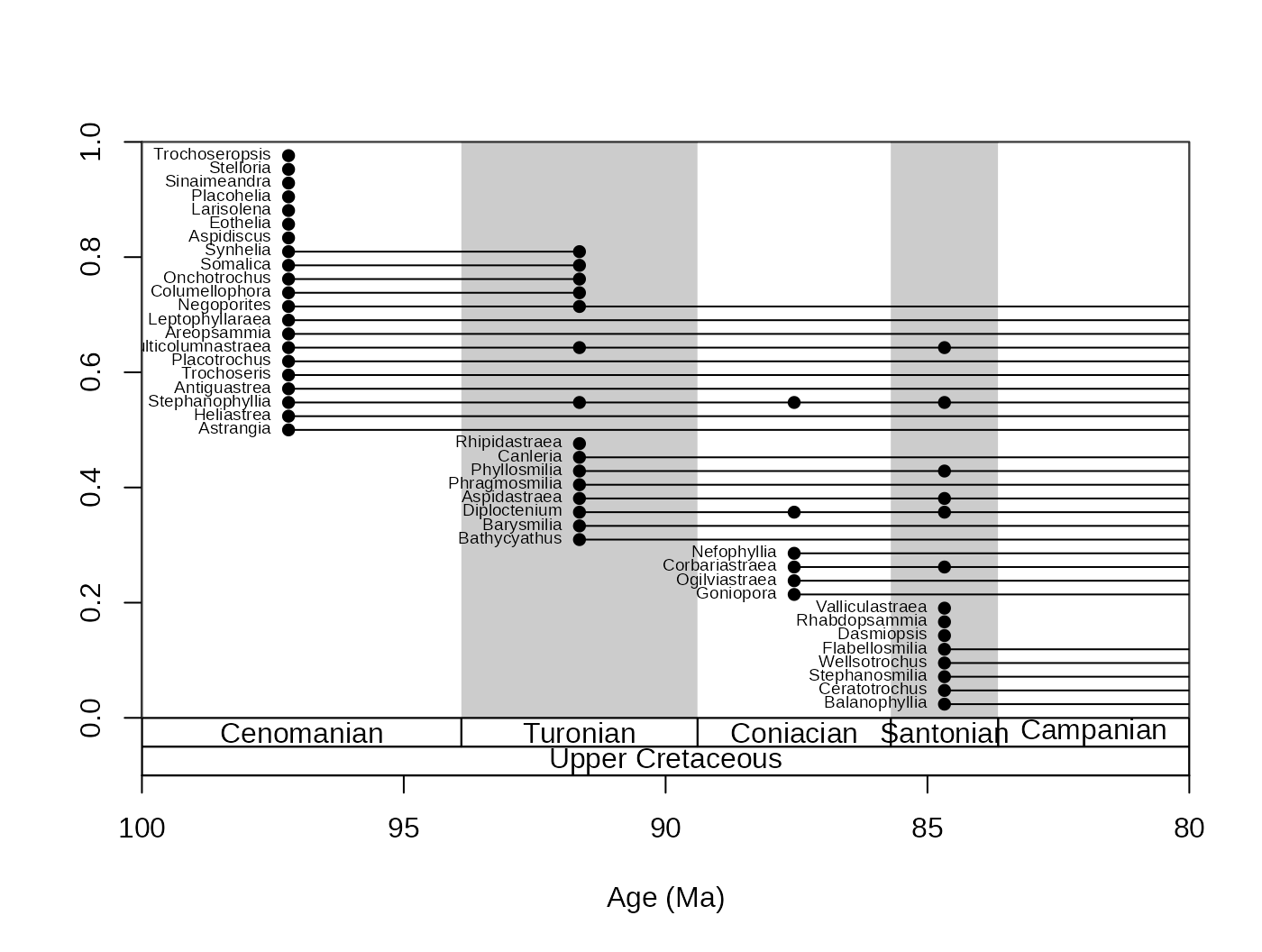
This function can also plot the taxa by groups. Here is the same plot, but by separating the taxa based on symbiotic status:
tsplot(stages, shading="stage", boxes=c("stage","series"),xlim=c(100,80))
ranges(fossils, tax="genus", bin="mid", labs=T,
labels.args=list(cex=0.6), filt="orig", occs=T, group="ecology")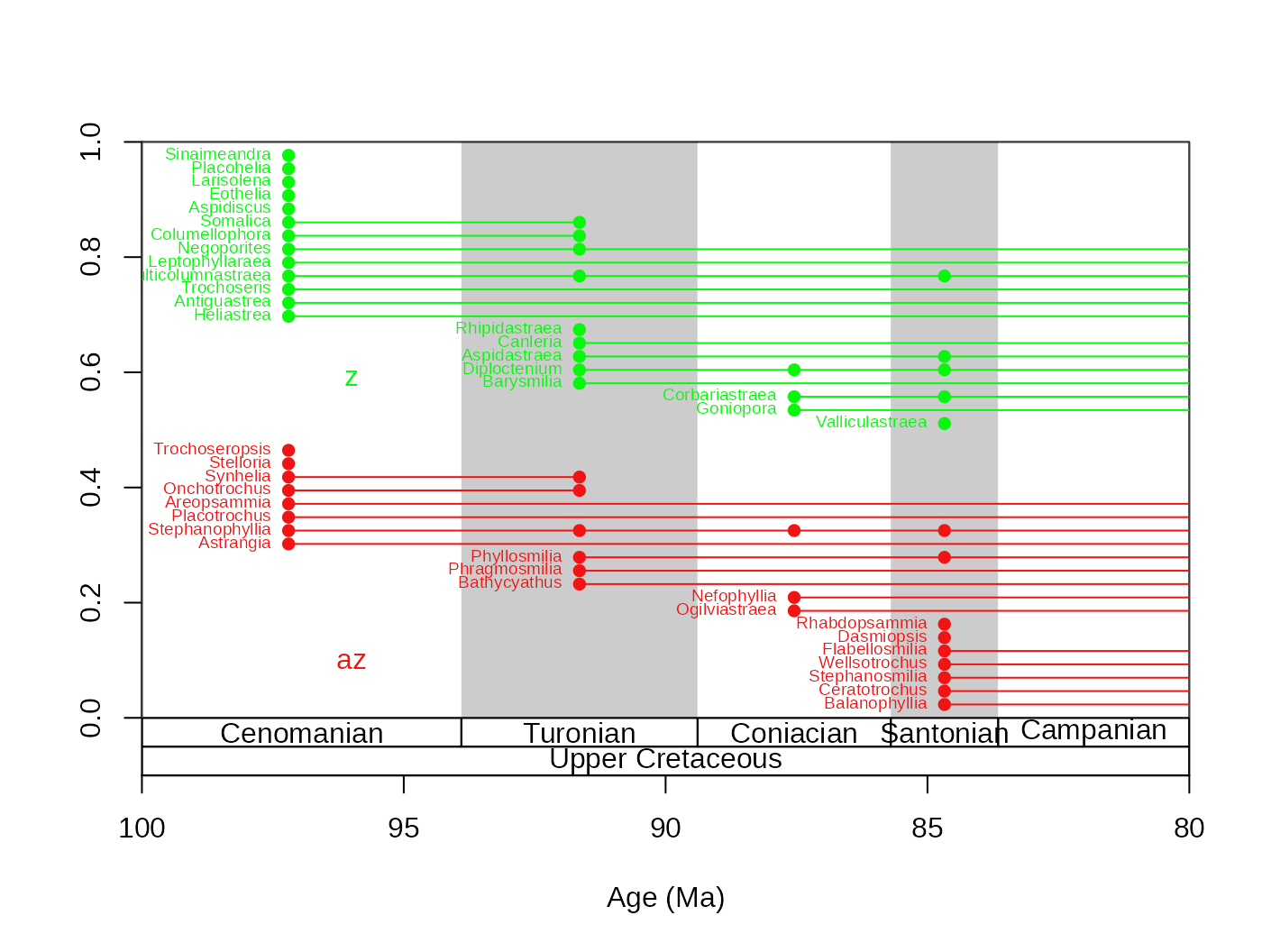
You can also plot survivorship curves. The function
survivors() calculates the proportions of survivors from
every bin to all the remaining bins.
surv <- survivors(corals, bin="stg", tax="genus")You can plot these values for every cohort to get survivorship
curves. As these curves can be thought of as products of an exponential
decay, it is customary to log the y axis, which you can do with adding
log="y" to the arguments of the main plot()
function in plot.args:
# time scale plot
tsplot(stages, shading="series", boxes="sys",
xlim=c(260,0), ylab="proportion of survivors present",
ylim=c(0.01,1),plot.args=list(log="y"))![]()
Then the curves can be plotted for every column with
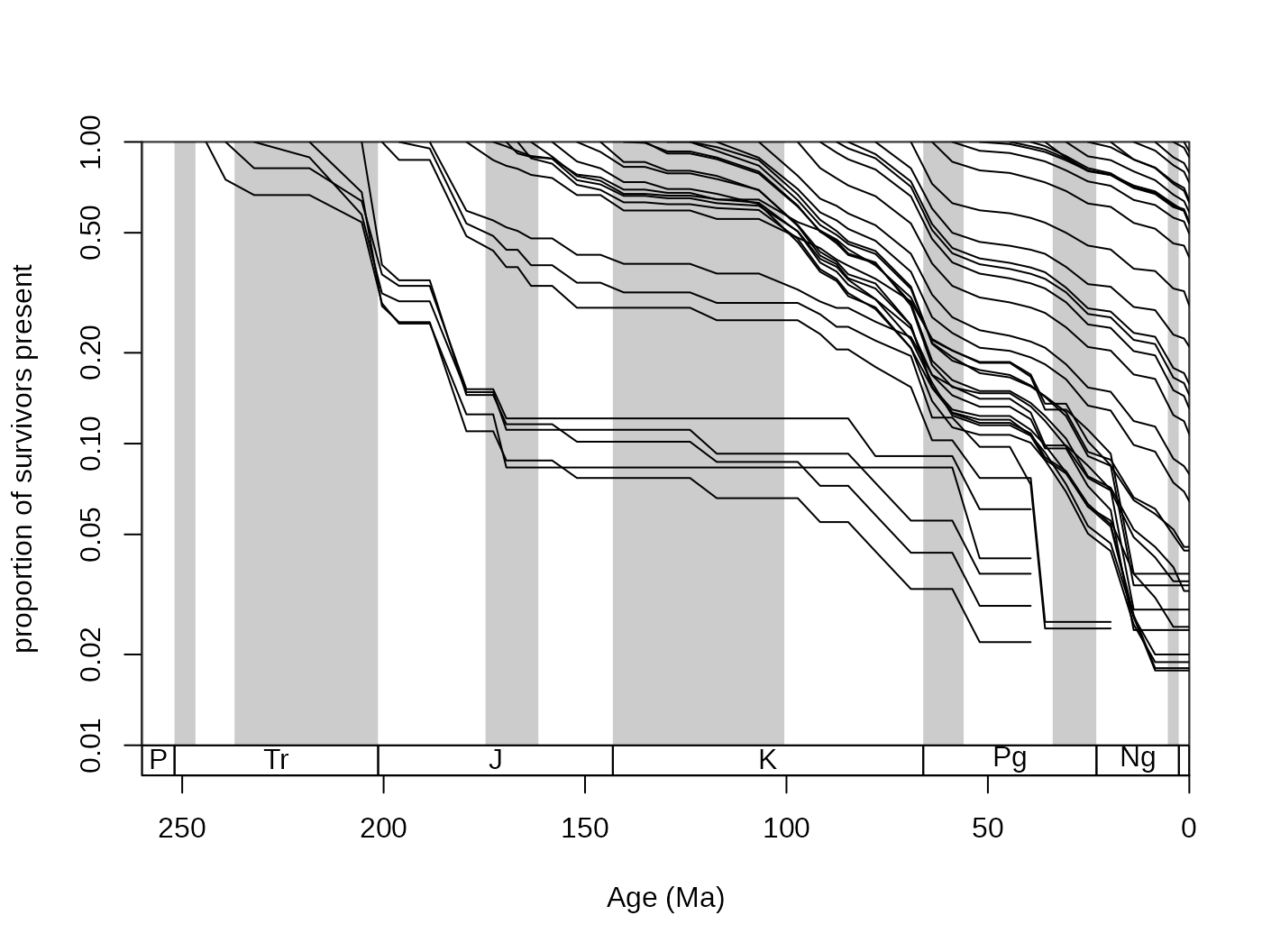
The values shown here are also called ‘forward’ survivorship
proportions, but you can also plot the ‘backward’ survivorships to see
how certain cohorts emerge over geologic time, by setting the
method argument of the survivors() function
appropriately:
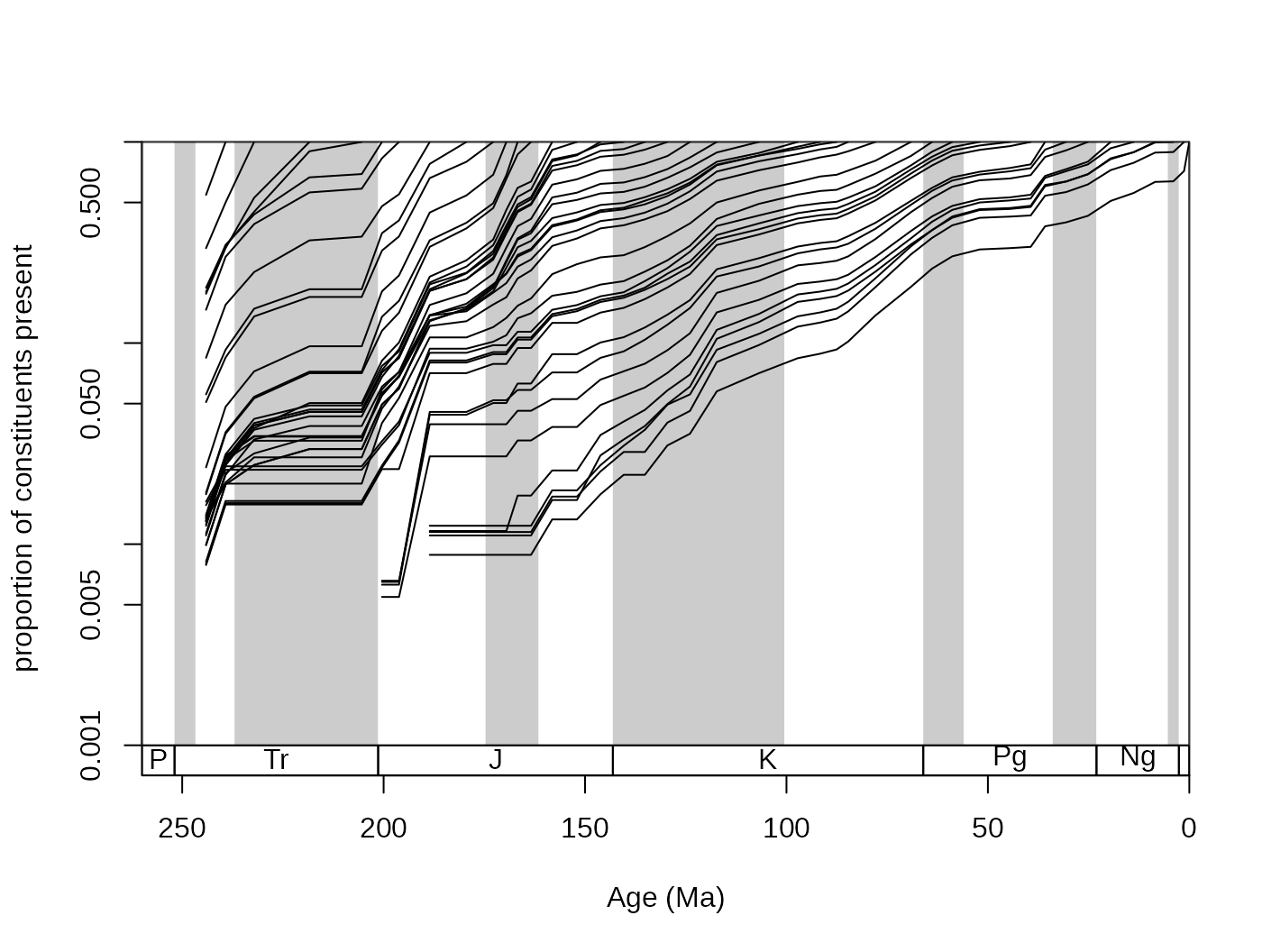
Large synchronous changes in these curves represent times where major changes happened in the history of the group. Major extinctions are apparent on the forward survivorship, whilst major origination episodes show up on the backward survivorship curves. However, ways that are more robust exist to quantify the factors that influence diversity.
3.2. Sampling parameters
3.2.1. Basic descriptors
The patterns of the data are constrained by sampling processes. These can have a direct influence on the patterns of diversity dynamics and therefore should be taken into consideration when the conclusions are drawn from the data.
The sumstat() function calculates basic sampling metrics
that characterize the entire dataset.
samp <-sumstat(fossils, tax="genus", bin="stg",
coll="collection_no", ref="reference_no", duplicates=FALSE)
samp## bins occs taxa colls refs gappiness
## 1 41 23229 760 5444 1203 0.5818335This includes the total number of occurrences, collections,
references and statistics of gappiness. You can also calculate most of
these basic sampling metrics for every bin with the
binstat() function.
samp <-binstat(fossils, tax="genus", bin="stg",
coll="collection_no", ref="reference_no")## The database contains duplicate occurrences (multiple species/genus).The message above indicates that multiple entries of the same genus
are present in the same collections. As this is a species-level
occurrence dataset, this is understandable. By default, these entries
are omitted from the calculations above, but you can toggle this
manually by setting the duplicates argument of binstat()
accordingly (TRUE will keep the duplicates,
FALSE will omit them – without notification).
The function calculates the basic bin-wise statistics that do not
require multi-bin pattern recognition (for instance for Alroy’s (2008)
three-timer sampling completeness, which is output by the
divDyn() function). Optionally, additional metrics
implemented in the indices() function can be applied to
information in every bin, by adding indices=TRUE to the
function call.
samp <-binstat(fossils, tax="genus", bin="stg",
coll="collection_no", ref="reference_no", indices=TRUE)## The database contains duplicate occurrences (multiple species/genus).
colnames(samp)## [1] "stg" "occs" "colls" "refs" "SIBs" "occ1"
## [7] "occ2" "ref1" "ref2" "u" "chao1occ" "uPrime"
## [13] "chao1ref" "richness" "shannon" "hill2" "dominance" "squares"
## [19] "chao2" "SCOR"All results of the output of this function can be plotted then in a
straightforward way, by referring to the elements of the data frame. For
instance, the occs element contains the number of sampled
occurrences and coll refers to the number of
collections:
oldPar <- par(mar=c(4,4,2,4))
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="number of occurrences", ylim=c(0,3000))
lines(stages$mid[1:94], samp$occs)
# the collections (rescaled, other axis)
lines(stages$mid[1:94], samp$colls*5, col="blue")
axis(4, col="blue",col.ticks="blue",col.axis="blue",
at=seq(0,3000,500), labels=seq(0,600,100))
mtext(4, text="number of collections", col="blue", line=2)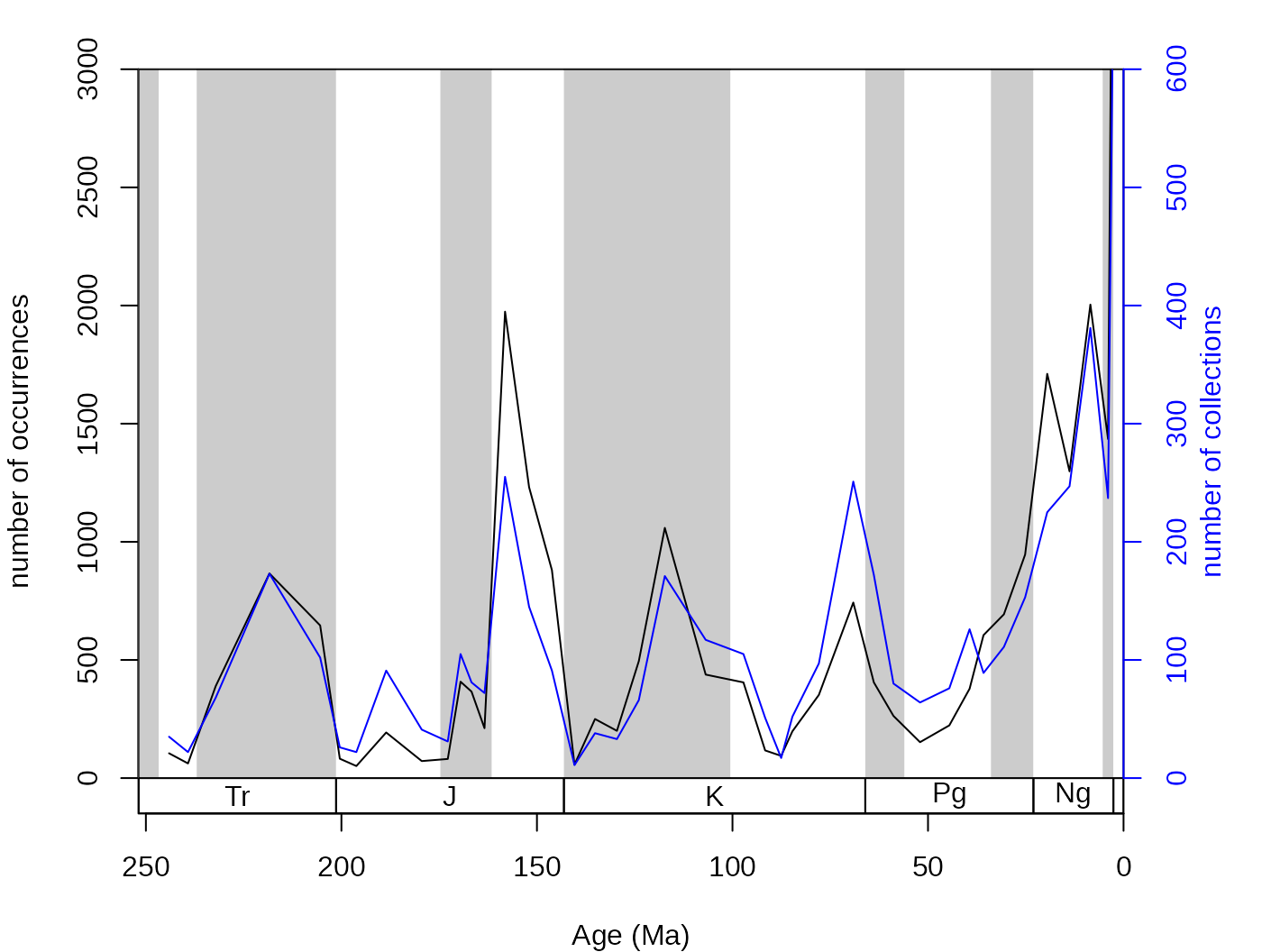
par(oldPar)The binstat() function also calculates other basic
statistics such as the numbers of references, sampled taxa,
single-collection taxa, single-reference taxa, double collection and
double reference taxa along with the sampling coverage estimator Good’s
u (1953), the coverage estimator suggested by Alroy (u’
, 2010) that is based on the number of single-reference taxa.
3.2.2. Plotting of counts and proportions
You can trace the trajectory of the occurrences that have different
attributes through the bins by using the parts() function.
This requires only two arguments: vector of bin identifiers and a vector
that contains the categories entries. The bin identifier also determines
the coordinates along the independent variable (time), so the numerical
bin entries have to be replaced by the age estimates.
# numerical ages, as bins
fossils$stgMid <- stages$mid[fossils$stg]
#plotting
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="number of occurences", ylim=c(0,3000))
parts(fossils$stgMid, fossils$bath)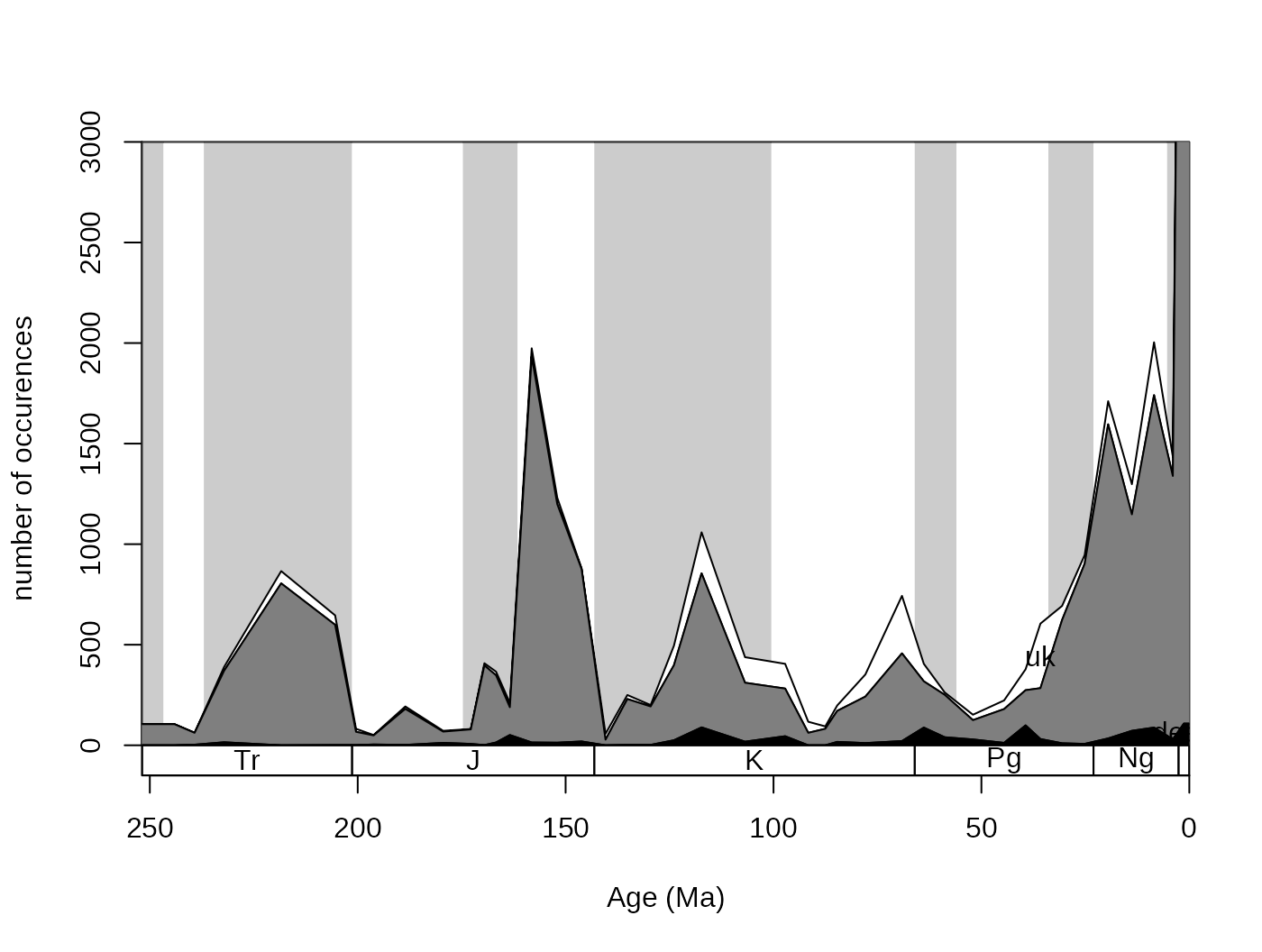
If you check out ?parts, there will be additional
examples using artificial data. In the default case, the category names
are plotted where they are the most abundant. This plot can be even
nicer if you use opacity (RGBA values for colors) and adding a proper
legend.
cols <- c("#FF0000AA", "#00FF00AA", "#0000FFAA")
# reorder too
reord <- c("shal","deep","uk")
plotnames <-c("shallow", "deep", "unknown")
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="the number of occurrences", ylim=c(0,3000))
parts(fossils$stgMid, fossils$bath, col=cols, ord=reord, labs=F)
legend("topleft", inset=c(0.01, 0.01),
legend= plotnames, fill=cols, bg="white")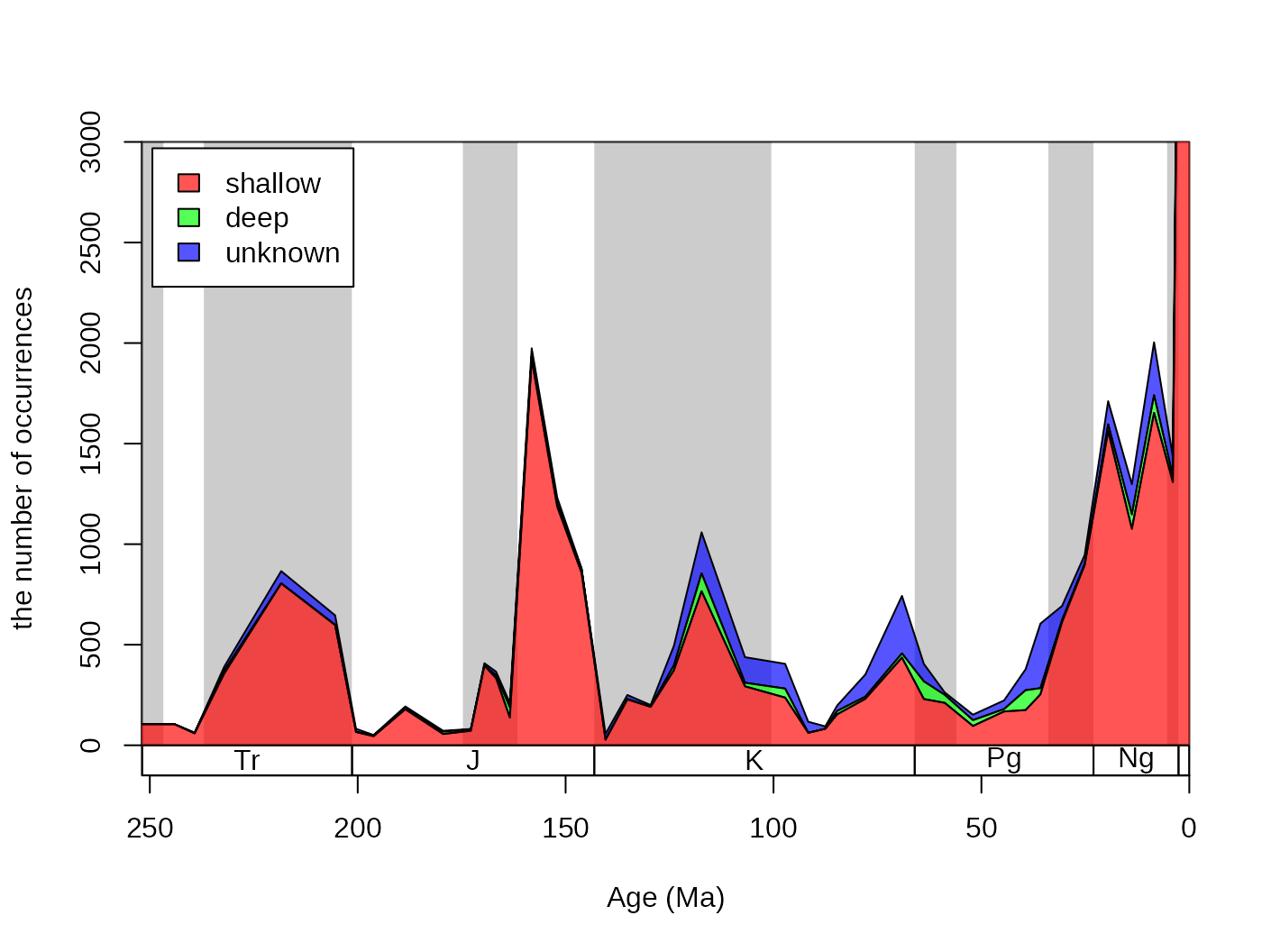
The dominance of shallow occurrences are even more apparent with
proportions. You can use the parts function to plot these,
rather than the counts, by adding prop=T to the function
call:
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="proportion of occurrences", ylim=c(0,1))
parts(fossils$stgMid, fossils$bath, prop=T, col=cols, ord=reord, labs=F)
legend("bottomleft", inset=c(0.01, 0.1),
legend= plotnames, fill=cols, bg="white")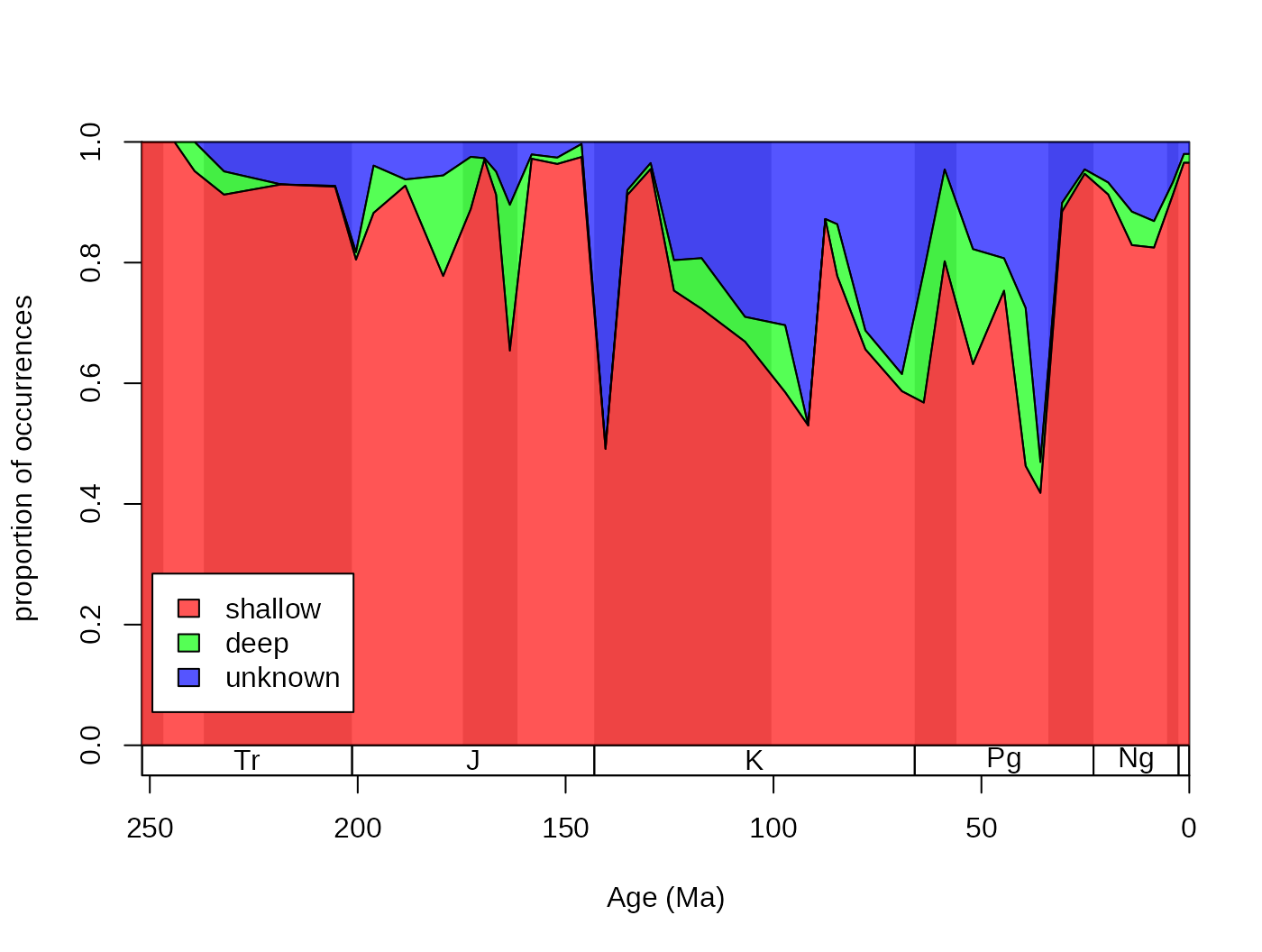
3.2.3. Single-collection and single reference taxa
Single-interval taxa were thought to be byproducts of temporary
increases of sampling intensity (Foote and Raup, 1996) in range-based
datasets such as Sepkoski’s compendium (2002). This phenomenon urged
researchers to develop metrics that ignore these taxa in calculating
metrics of biodiversity and evolution. Single-collection taxa can be
quickly omitted from the datasets by using the omit()
function. This will return a logical vector, indicating the rows that
should be omitted. The om argument can be set either to
omit the single-collection occurrences (om="coll").
Single-reference taxa can be omitted in the same way, although the term
needs additional clarification if multiple bins exist. Taxa that were
only described in a single reference (om="ref"). Some taxa
appear in multiple bins and in each of them they are described by only
one reference. These you can omit with om="binref".
omitColl <- omit(corals, tax="genus", om="coll", coll="collection_no")
omitRef <- omit(corals, tax="genus", om="ref", ref="reference_no")
omitBinref <- omit(corals, bin="stg", tax="genus", om="binref", ref="reference_no")
# the conserved number of occurrences will be
sum(!omitColl)## [1] 29583
sum(!omitRef)## [1] 29489
sum(!omitBinref)## [1] 291054. Raw diversity dynamics
The main calculations of the package are contained in the
divDyn() function. This function calculates patterns of
taxon occurrences and stratigraphic ranges to derive estimates over time
for richness, origination/extinction rates and sampling probabilities.
In order to calculate these you only need an occurrence table with
variables including the taxon names (tax) and the time
identifiers (bin). The rest of the information in the table
will not be used by the function.
ddFirst<-divDyn(corals, bin="stg", tax="genus", noNAStart=TRUE)The output of the divDyn() function is a
data.frame class object that contains values for each bins
in rows. The first column (bin variable) contains the bin
identifiers. The rest of the variables are explained in the
documentation.
4.1. Diversity (taxonomic richness) with discrete time input
You read this because you have interests in the changes of diversity.
Calculating time series of diversity does not require additional action
than running the basic divDyn() function.
4.1.1.Taxon counts
All calculations in this function depend on patterns of occurrences
in the bin/taxon matrix. The numbers of taxa that belong to different
categories form the basis of the metrics. These categories are explained
by Foote (1999) and Alroy (2014) and are available in the help file
?divDyn.
4.1.2. Different metrics of richness
Calculating time series of diversity does not require additional
action than running the basic divDyn() function. In this
package, among the traditional range-based methods the range-through
(variable divRT) and the boundary-crosser
(divBC) diversities are implemented. Plotting can be also
facilitated by matching the indices of the diversity values in the
variables and their numbers. If the bin numbers are positive integers
this is turned on by setting the noNAStart argument to
FALSE, which is the default. This change results in
NAs and zeros in the final table in rows, which bins are
not sampled.
# metrics
ddRec <-divDyn(corals, bin="stg", tax="genus")
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="range-through richness (diversity)", ylim=c(0,250))
# lines
lines(stages$mid, ddRec$divRT, col="black", lwd=2)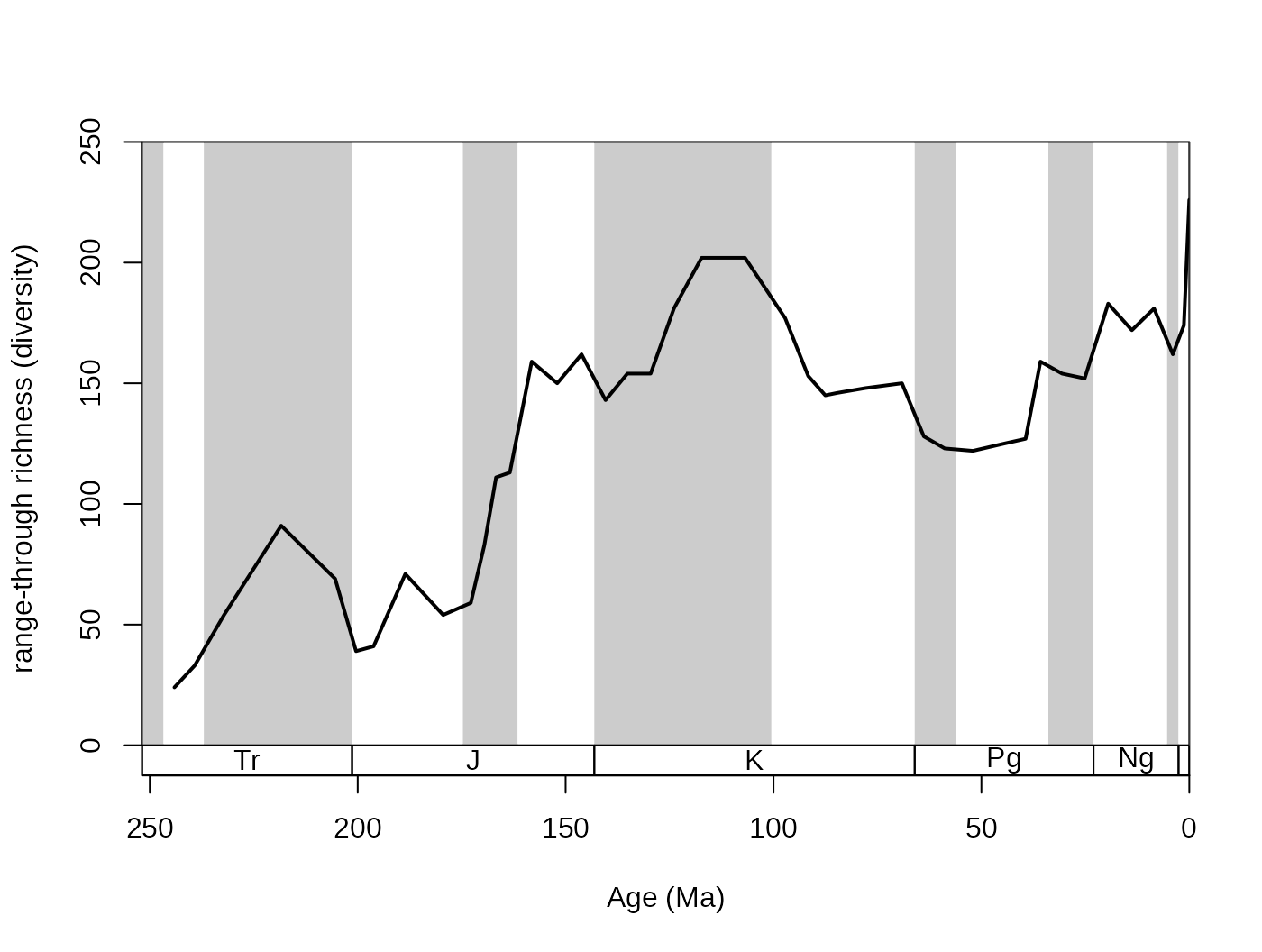
This plot shows the trajectory of coral diversity over the
post-Permian interval, using raw data and the range-through diversity
metric. Besides the 0 values at the start of the time series (no sampled
corals), you might also notice a sharp increase of richness in the
latest intervals. This could be the result of an effect called the ‘Pull
of the Recent’. The ‘Pull of the Recent’ (Raup, 1979) is a smearing
phenomenon that arises when range-based methods are calculated from
datasets where sampling probability changes abruptly. As we know the
Recent much better in comparison to other intervals (sampling
probability is much higher, around one), the number of ranges that
connect first occurrences of taxa to the extant time interval
(stg == 95) is much higher than those that link two
non-Recent time intervals. The results in spur of diversity as the
Recent is approached. On the other hand, omitting the recent interval
(effectively decreasing sampling probability to 0) leads to edge effects
that result in the opposite phenomenon. The discrepancy between these
two approaches can be visualized by the omission of the recent
‘occurrences’:
# metrics
dd <-divDyn(fossils, bin="stg", tax="genus")
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="richness (diversity)", ylim=c(0,250))
# lines
lines(stages$mid, ddRec$divRT, col="black", lwd=2)
lines(stages$mid[1:94], dd$divRT, col="blue", lwd=2)
# legend
legend("topleft", legend=c("with recent", "without recent"), col=c("black", "blue"), lwd=c(2,2), bg="white")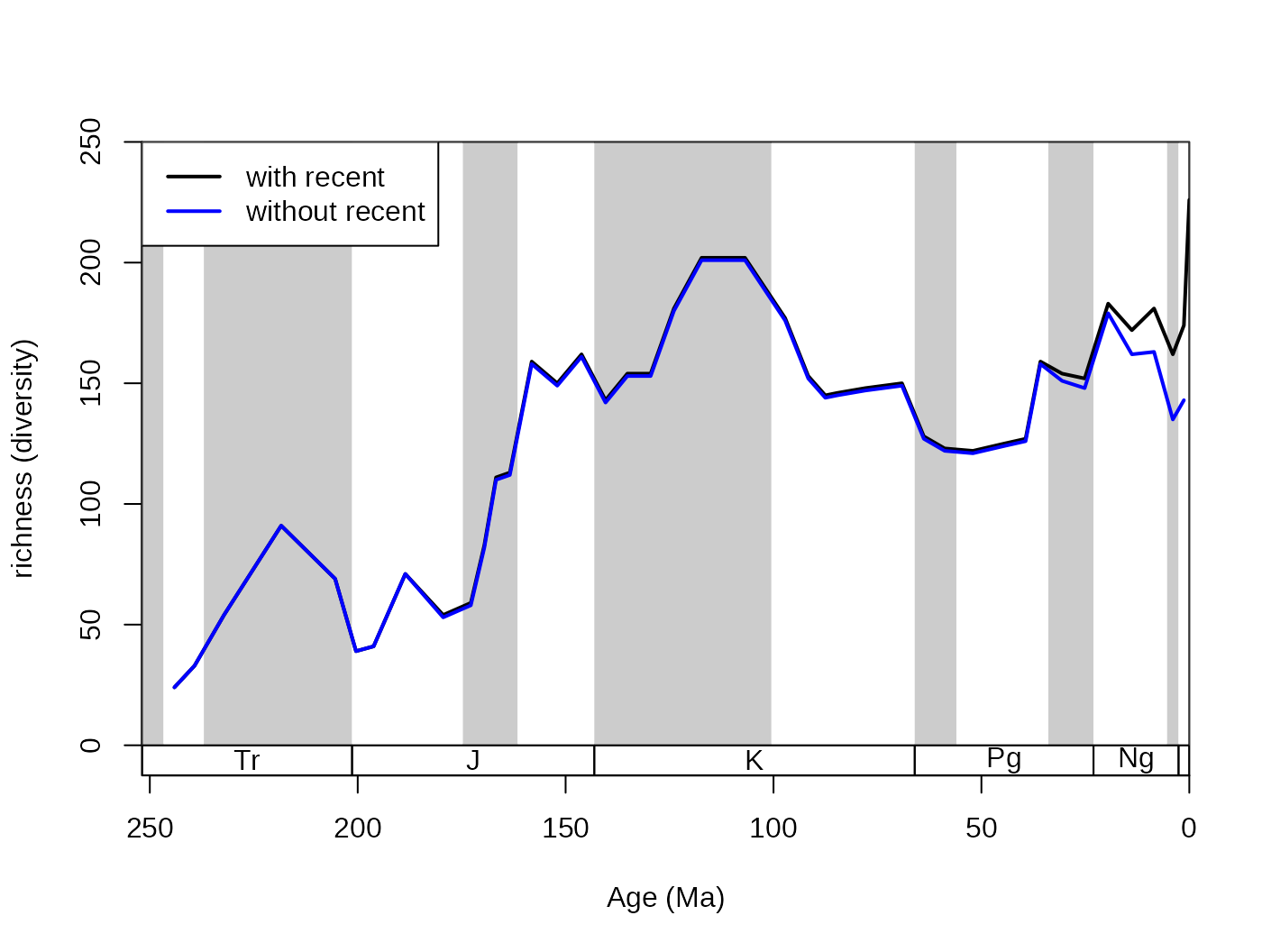
Over the last couple of decades, a number of metrics were proposed to
efficiently express diversity given such distorting effects of
incomplete sampling and the discretized time dimension. As these more
straightforward approaches that use range interpolations have known
issues, occurrence datasets, allow the calculation of more direct
estimators, such as sampled-in-bin (SIB, variable divSIB)
diversities.
# metrics
dd <-divDyn(fossils, bin="stg", tax="genus")
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="richness (diversity)", ylim=c(0,250))
# lines
lines(stages$mid[1:94], dd$divRT, col="red", lwd=2)
lines(stages$mid[1:94], dd$divBC, col="blue", lwd=2)
lines(stages$mid[1:94], dd$divSIB, col="green", lwd=2)
# legend
legend("topleft", legend=c("RT", "BC", "SIB"),
col=c("red", "blue", "green"), lwd=c(2,2,2), bg="white")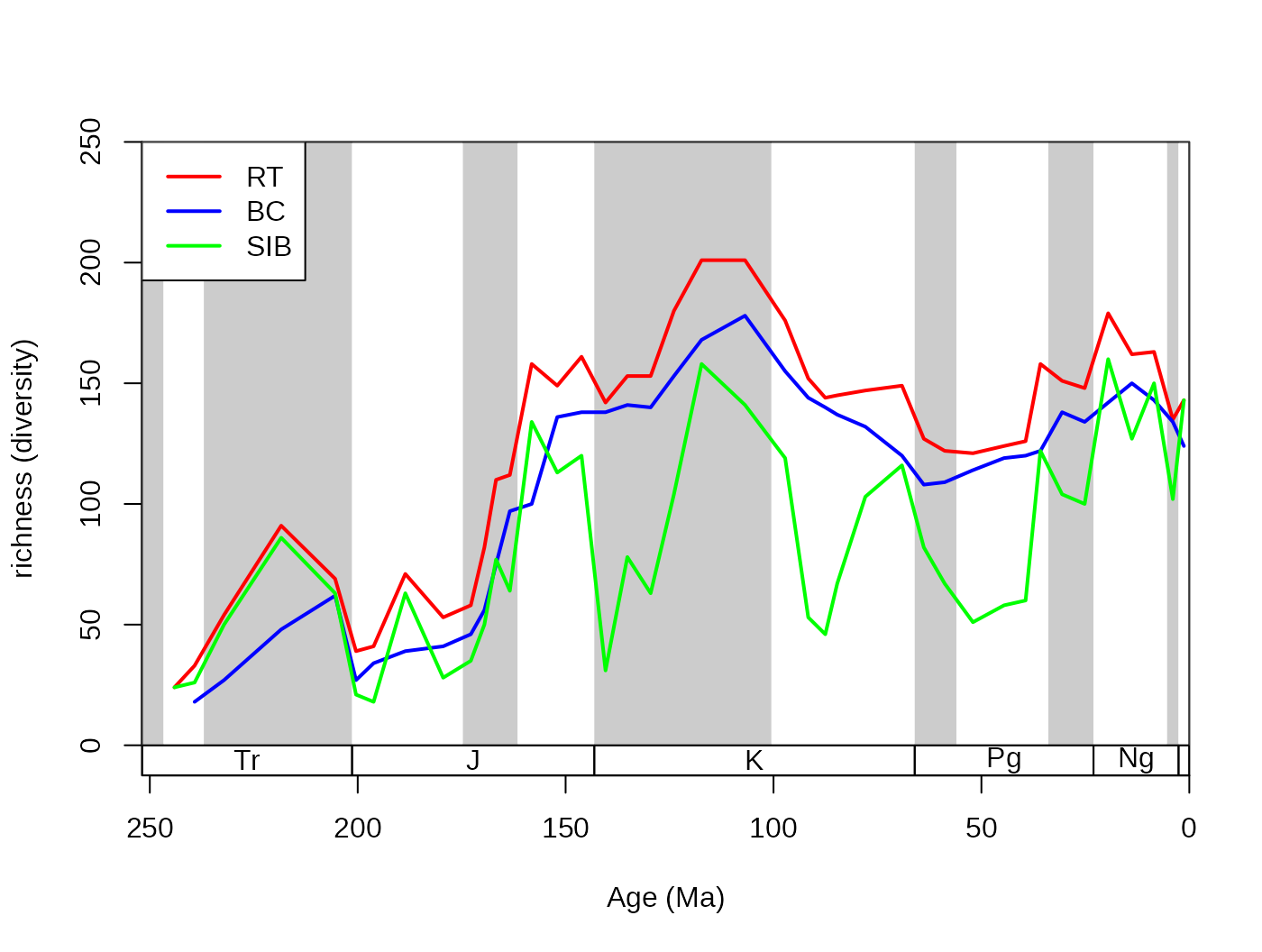
Although range-interpolation does not bias the ‘SIB’ metric, it is
more affected by changes of sampling intensity sampling. The three-timer
sampling completeness is an effective expression of changes in sampling
(samp3t variable):
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="three-timer sampling completeness")
# lines
lines(stages$mid[1:94], dd$samp3t, col="black", lwd=2)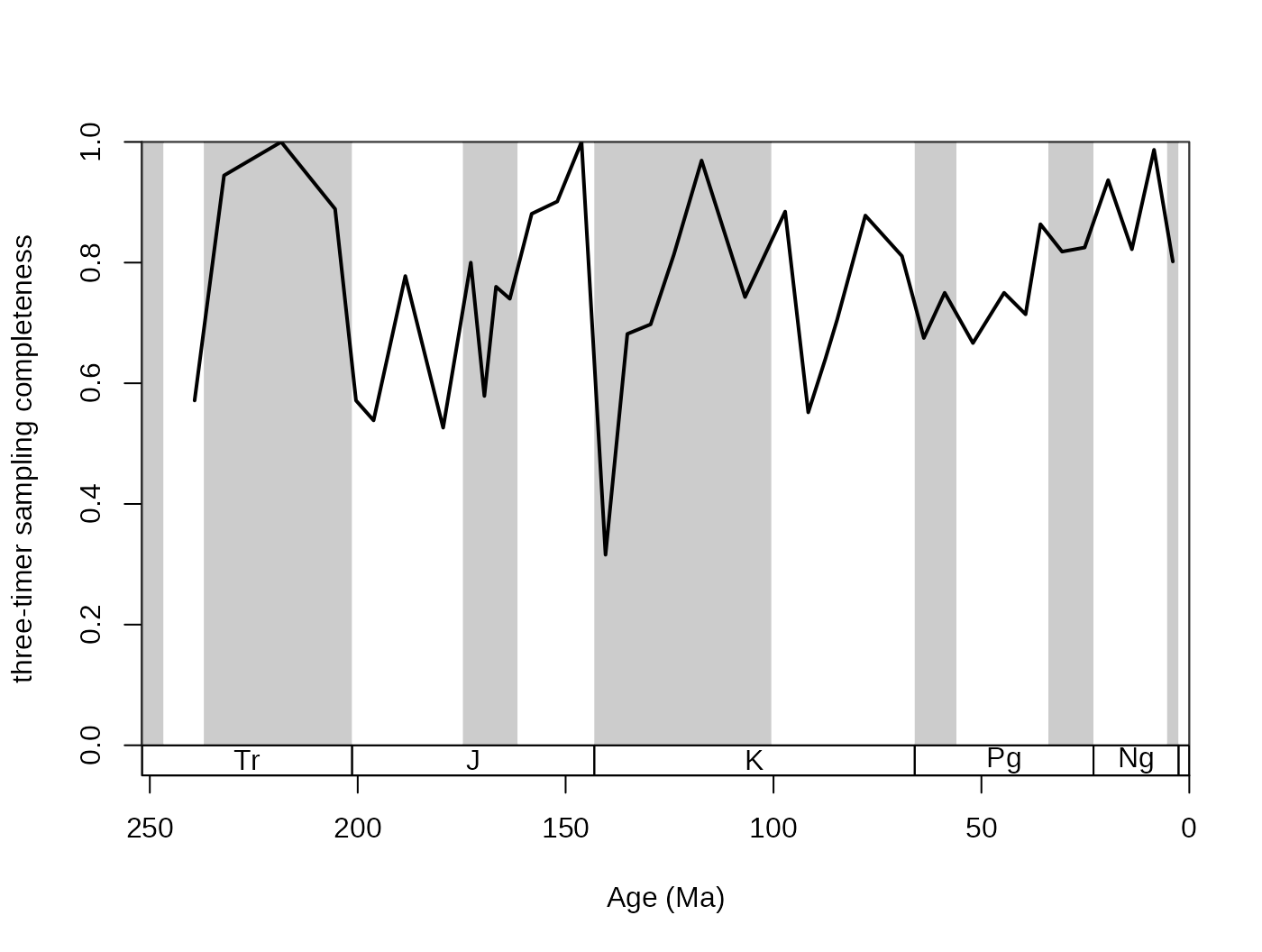
The SIB series can be partially corrected by the three-timer sampling-completeness (Alroy §ref ). Although this can be a convenient correction it also increases the estimation error. Nevertheless, this is the least biased estimator for diversity.
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="three-timer sampling completeness", ylim=c(0,250))
lines(stages$mid[1:94], dd$divSIB, col="black", lwd=2)
lines(stages$mid[1:94], dd$divCSIB, col="blue", lwd=2)
# legend
legend("topleft", legend=c("SIB", "corrected SIB" ),
col=c("black", "blue"), lwd=c(2,2), bg="white")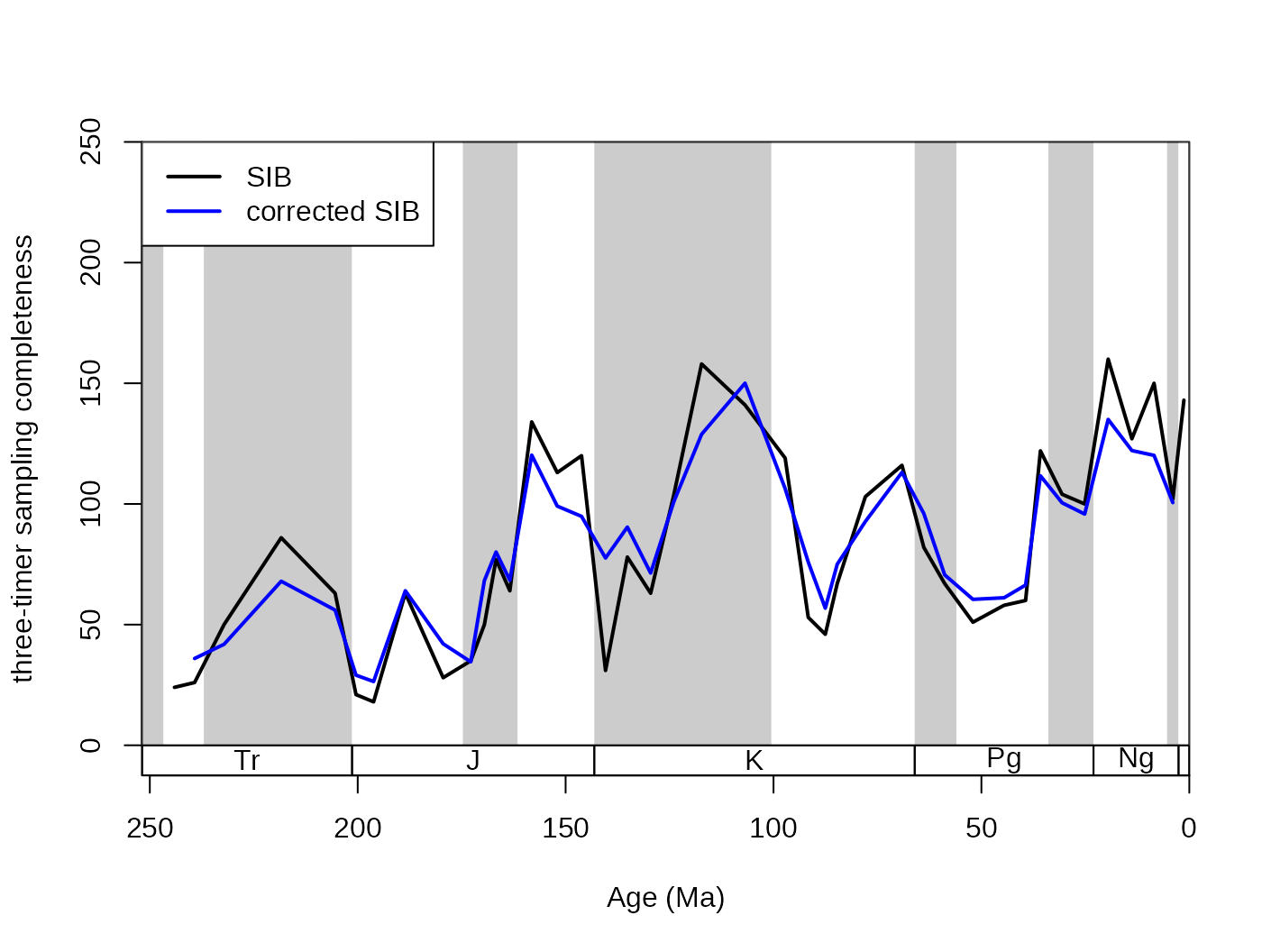 The
The omit() function is also embedded in the
divDyn() function, so that the metrics are calculated after
the omission of poorly sampled taxa (switched off by default). You can
add this filter, by adding (the appropriate om arguments to
the divDyn() function call.
4.2. Taxonomic rates with discrete time input
Analyzing time series of originations and extinctions helps us to
describe not just how diversity changed over time, by also why it
changed. With taxonomic rates we can decompose changes in diversity to
the relative contribution of cladogenetic processes (i.e. origination,
the birth of a new taxon) and that of the disappearance of taxa
(i.e. extinction, the death of a taxon). In the discrete time model, the
most straightforward way to express these contributions is through
simple exponential decay models (Raup, 1985). For convenience, these
variables are named as extTYPE and oriTYPE for
extinction and origination rates, respectively.
4.2.1. Different metrics of turnover
In the package, the proportional rates (extProp and
oriProp) and per capita rates (extPC and
oriPC) of Foote (1999), the three-timer rates
(ext3t and ori3t) of Alroy (2008), their
corrected counterparts (extC3t and oriC3t),
the gap-filler rates (extGF and oriGF) of
Alroy (2014) and the improved second-for-third (ext2f3 and
ori2f3) of Alroy (2015) are provided.
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="extinction rates", ylim=c(0,2))
lines(stages$mid[1:94], dd$extPC, col="black", lwd=2)
lines(stages$mid[1:94], dd$extGF, col="blue", lwd=2)
lines(stages$mid[1:94], dd$ext2f3, col="green", lwd=2)
# legend
legend("topright", legend=c("per capita", "gap-filler", "second-for-third"),
col=c("black", "blue", "green"), lwd=c(2,2,2), bg="white")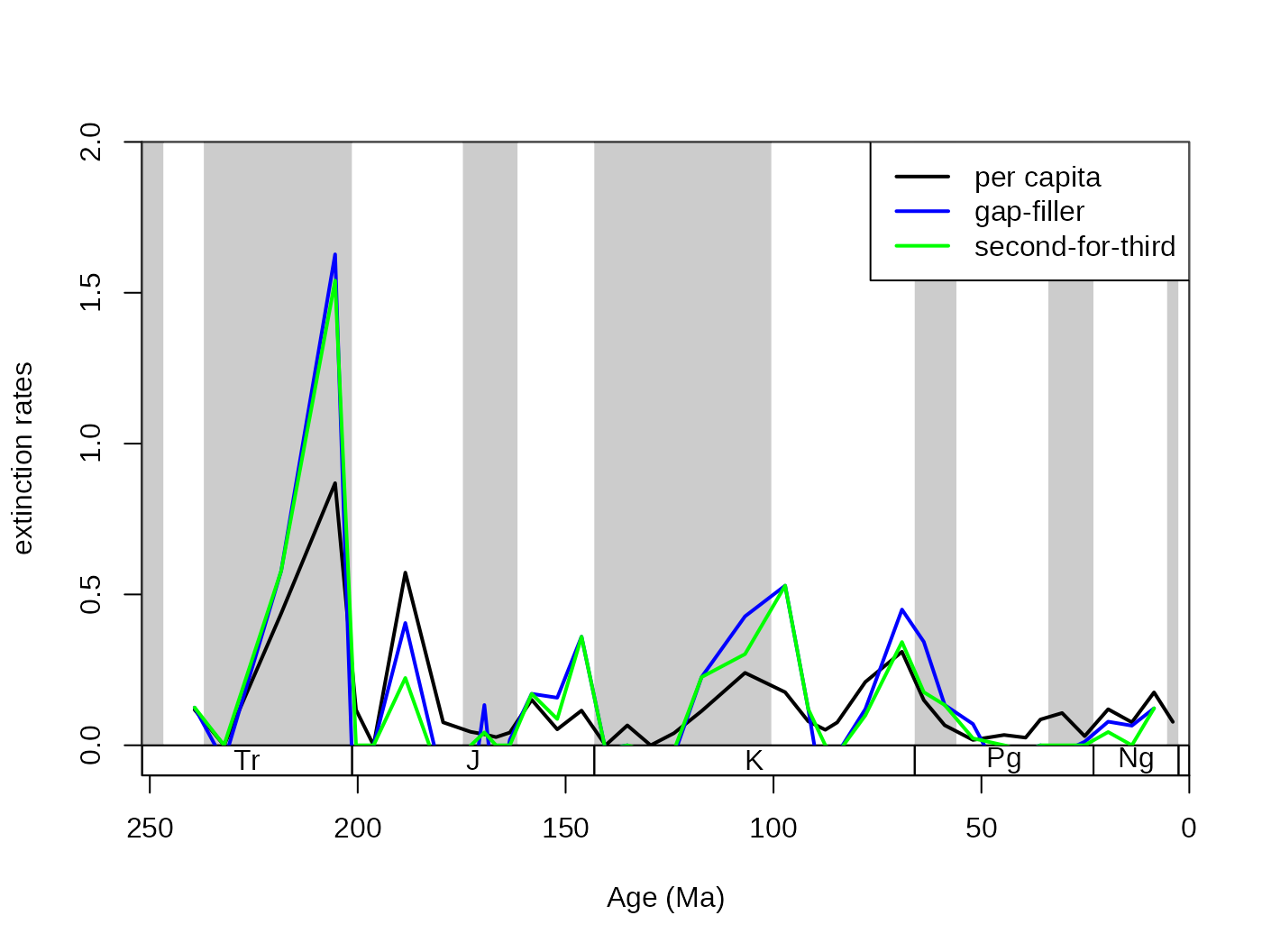
The documentation of the function (type in console ?divDyn) contains the formulas for the calculation of the rates. With heterogeneous, incomplete sampling the metrics have different properties that can be summarized as follows:
Proportional ‘rates’: These metrics express what proportion of the cohort of taxa disappears until the new interval.
The per capita rates of Foote (1999) use the range-through assumption to establish ranges for the taxa in the dataset. The rate value expresses what proportion of the taxa decayed until the end of the interval. The method is biased by the Signor-Lipps effect and edge effects (Foote 2000).
The ‘three-timer’ and ‘corrected three-timer’ rates (Alroy, 2008) are different estimators of the per capita rates but will converge on them when sampling tends to completeness. They use moving windows around the focal interval to select data that is more relevant to the focal interval. This metric is unbiased by the Signor-Lipps and edge effects. The three-timer sampling completeness can be used to correct this metric, but it will not improve its susceptibility to random sampling error.
The ‘gap-filler’ rates (Alroy, 2014): gap-filler extinction rates are improved versions of the three-timer rates, using four-bin moving windows instead of three. This makes the rates less resistant to random error (more taxa are recognized by the categorizing procedure) and are an improvement on the three-timer rates. The method sometimes results in the seemingly nonsensical negative extinction rates, when the true extinction rates are near 0.
The ‘second-for-third’ rates (Alroy, 2015) are further improvement of ideas the three-timer and gap-filler rates represent. By adding an algorithmic approach the frequency of negative extinction rates are further decreased. Extinction and origination proportions described by Alroy (2015) can also be used (
E2f3andO2f3, respectively).
4.2.2. Selectivity testing for the per capita rates
Although extinction and origination rates can provide information about events and background processes on their own, sometimes it is more useful to assess them in comparison between taxa. The most frequent way of doing this is by splitting a group to subsets and comparing the patterns of turnover in certain intervals between the two groups. In the case of selectivity testing, an ecologically, taxonomically important grouping variable indicates that one of the groups has a higher turnover at a specific point in time. For the sake of convenience in hypothesis testing for selectivity for one state of the variable (e.g. heavily calcified), the available methods assess selectivity between two groups.
The general problem of extinction selectivity is the error estimation of the rate values. More data (e.g. more taxa) will lead to more reliable estimates while, splitting the same dataset to two subsets will lead to higher uncertainty in the rate estimates for the two rate values in the selected interval. The selectivity testing incorporates two aspects: (1) the difference between the two rate values, (2) and whether this distance is meaningful or not. In practice, these two criteria can be summarized by whether it is more supported by the data to describe two rate values, or is it better founded to describe just one. These two alternatives are sometimes called as single-rate model and the dual-rate model (Kiessling and Simpson, 2011; Kiessling and Kocsis, 2015). In the case that a dual rate model is better supported, that means that the difference between the two values of rates is statistically meaningful, and the extinction or origination event selectively affected the group with the higher value. For example, here are the raw origination rates of corals, plotted for the az and the z group separately and together:
# split by ecology (az = azooxanthellate, z = zooxantehllate)
z<- fossils[fossils$ecology=="z",]
az<- fossils[fossils$ecology=="az",]
# calculate diversity dynamics
ddZ<-divDyn(z, tax="genus", bin="stg")
ddAZ<-divDyn(az, tax="genus", bin="stg")
# origination rate plot
tsplot(stages, boxes="sys", shading="series", xlim=54:95,
ylab="raw per capita originations")
lines(stages$mid[1:94], dd$oriPC, lwd=2, lty=1, col="black")
lines(stages$mid[1:94], ddZ$oriPC, lwd=2, lty=1, col="blue")
lines(stages$mid[1:94], ddAZ$oriPC, lwd=2, lty=2, col="red")
legend("topright", inset=c(0.1,0.1), legend=c("total", "z", "az"),
lwd=2, lty=c(1,1,2), col=c("black", "blue", "red"), bg="white")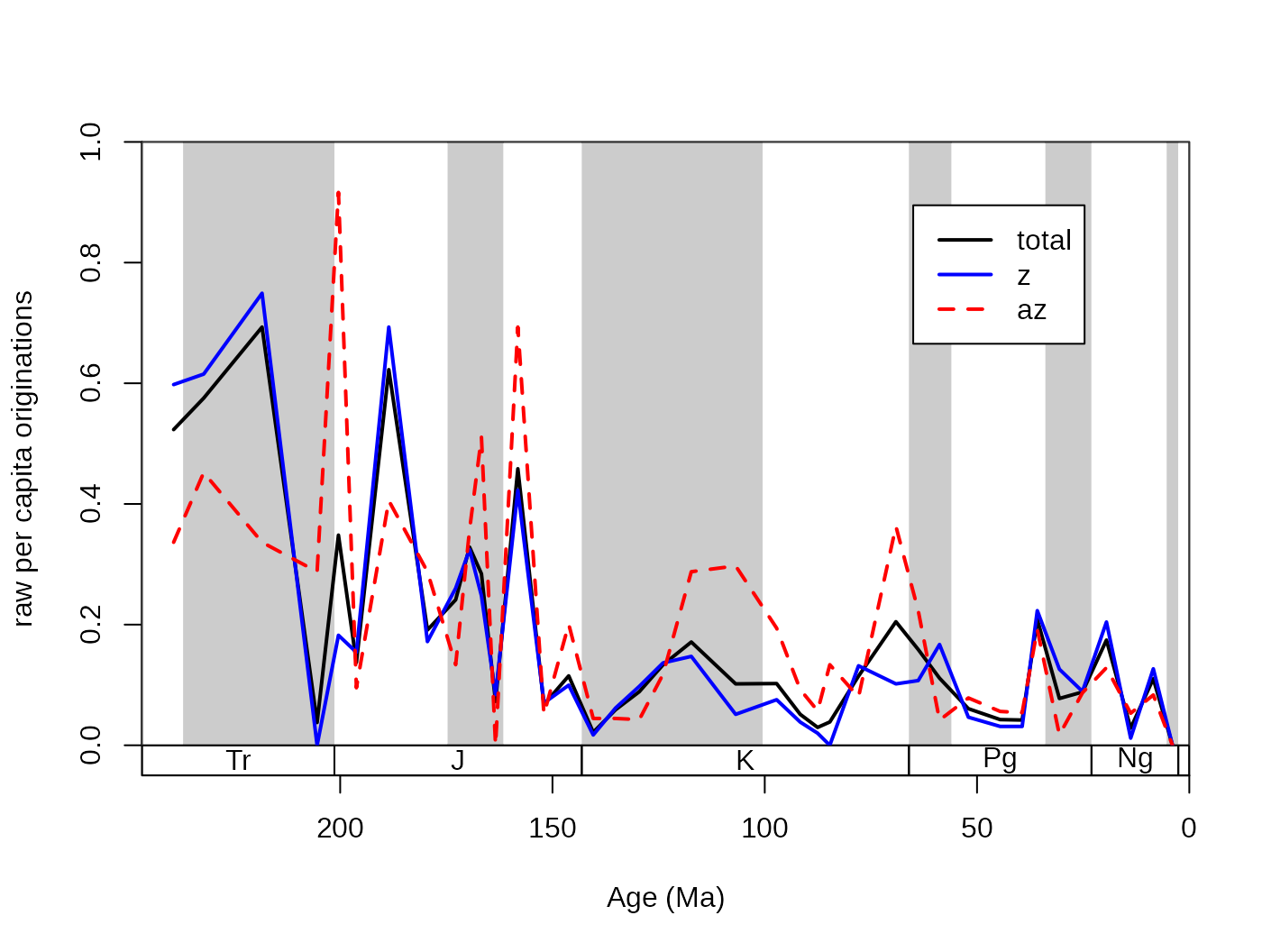
The question that arises in this case is whether some intervals can
be characterized by actually higher origination rates (for az
taxa in the Cretaceous), or is this just a sampling artifact. The
testing framework is implemented currently only for the per capita
extinction rates of Foote (1999) in the ratesplit function.
The function will take the occurrence dataset as an argument and will
calculate the rates values similarly to the divDyn
function. The function requires a separator variable for the selection
testing (sel) that will be used for splitting the dataset
into two (this column has to have two possible states i.e. binary code).
The implementation of the process for the symbiotic status is:
rs<-ratesplit(fossils, sel="ecology", tax="genus", bin="stg")
rs## $ext
## integer(0)
##
## $ori
## [1] 57 59 75 81The default output of this function by default is a list of two
vectors: ext and ori, extinction rates and
origination rates, respectively. Each vector contains the bin numbers
where the dual rate model is better supported than the single rate
model, where selectivity is plausible. In the example above, selectivity
for the extinctions is unlikely and it is supported for bin 57 (Norian),
59 (Hettangian), 75 (Albian) and 81 (Maastrichtian). The statistical
testing in this case is based on the number of taxa and can be performed
in two different ways, set with the method argument. The
less stringent binom method implements binomial testing,
with a significance value set with the alpha parameter that
defaults to 0.05.
rsBin95<-ratesplit(fossils, sel="ecology", tax="genus",
bin="stg", method="binom")
rsBin95## $ext
## [1] 62
##
## $ori
## [1] 57 59 75 79 81 83 88
rsBin90<-ratesplit(fossils, sel="ecology", tax="genus",
bin="stg", method="binom", alpha=0.1)
rsBin90## $ext
## [1] 62 80
##
## $ori
## [1] 57 59 65 74 75 76 79 81 82 83 88The more conservative approach is to use model selection criteria for
the testing of support for the single or dual rate
models, which involves calculating Akaike Information Criteria and
then Akaike weights. This is the default method
(method=AIC). The alpha argument in this case
depicts the minimum Akaike weight that serves as a threshold for the
dual rate model to be supported. As the ratio of these weights
represent likelihood ratios, by default it is set to 0.89 that roughly
represents 8 times higher likelihood for the dual rate model.
This can be toggled the way it is deemed useful for the question at
hand.
rsAIC0.5<-ratesplit(fossils, sel="ecology", tax="genus",
bin="stg", alpha=0.5)
rsAIC0.5## $ext
## [1] 57 66 80 81
##
## $ori
## [1] 57 59 65 74 75 76 81 82 83 88 91In the case above, the 0.5 alpha value indicates that
the dual rate model should be supported if it is more likely than the
single rate model.
This output, listing supported intervals, is useful for plotting the selectivity values that could be easily visualized by dots. You can add these to the plot of z and az coral origination rates by running:
# select rate values in the intervals of selectivity
selIntervals<-cbind(ddZ$oriPC, ddAZ$oriPC)[rs$ori,]
# which is higher? TRUE: AZ, FALSE: Z
groupSelector<-apply(selIntervals, 1, function(x) x[1]<x[2])
# draw the points
# for the AZ corals
points(
stages$mid[rs$ori[groupSelector]],
ddAZ$oriPC[rs$ori[groupSelector]],
pch=16, col="red", cex=2)
# for the Z corals
points(
stages$mid[rs$ori[!groupSelector]],
ddZ$oriPC[rs$ori[!groupSelector]],
pch=16, col="blue", cex=2)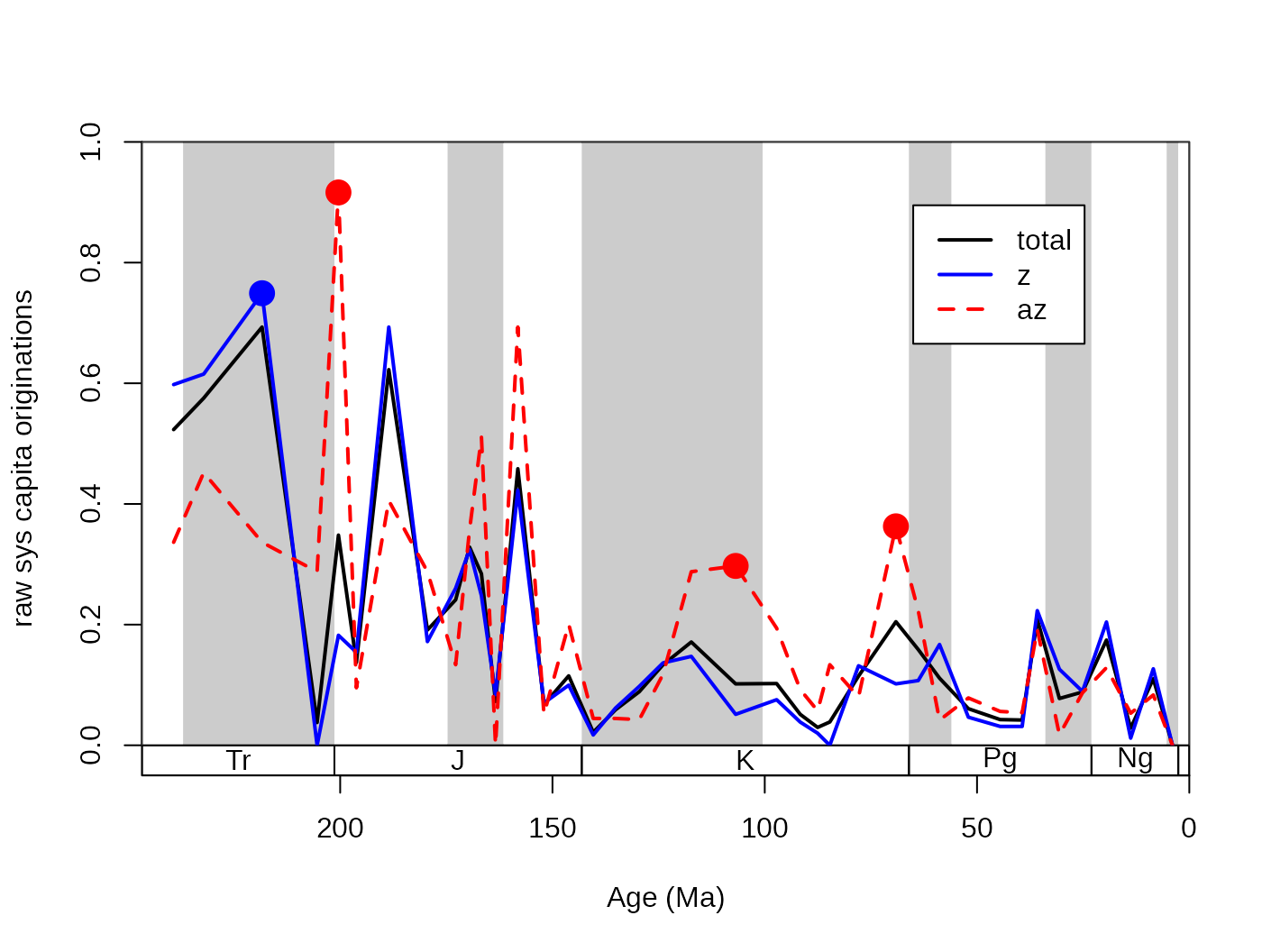 The
The combine method will employ both schemes for testing,
but with the default alpha levels, the binomial test would not limit the
set of selective intervals further than the one base on
AIC.
Naturally the ratesplit() function can return the
p-values of the binomial tests and the Akaike weights by
setting the output argument to "full" .
4.3. Diversity dynamics with age input
The added option to use a time variable that contains real values allows users to use different data to express the passing of time. This addition enables the application of the basic function to examples of high temporal resolution, where time is close to continuous, or samples in relative time such as expressed by meters within the section.
4.3.1. Using the bin argument – unique values code
intervals
In the case of the coral dataset, we can try this numerical binning approach by assigning the estimated age mid points to the occurrences. This is a very ‘dirty’ way of treating occurrence data as the assumed uncertainty of the age estimate is practically reduced to 0, and overlapping intervals that express uncertainty will have different assigned bins.
If this column is set as the bin argument, then
occurrences will be treated to represent the same time interval that
have the same value in this column. The entries will be ordered by the
function. Similarly to physical systems time is assumed to flow from
lower to larger values when it is provided as the
bin argument. This means that time
has to be reversed, otherwise extinctions become originations and vice
versa. You can toggle this by setting
revtime=TRUE.
ddIDbin <- divDyn(fossils, tax="genus", bin="mid_ma", revtime=TRUE)Please note that this is example is for illustration purposes only.
As there are too many unique entries in this bin variable,
with the corals, this resolution will be too high to produce potentially
meaningful patterns. Range-through diversities are possibly the closest
to reality with this method. With this type of binning, the first
variable of the divDyn output (name specified in
bin) contains the ages for the bins, which you can use for
plotting.
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="Diversity, range-through", ylim=c(0,300))
lines(ddIDbin$mid_ma, ddIDbin$divRT, col="black", lwd=2)
lines(stages$mid[1:94], dd$divRT, col="red", lwd=2)
legend("topleft", legend=c("unique mid entries", "stg stages"),
col=c("black", "red"), lwd=c(2,2), bg="white")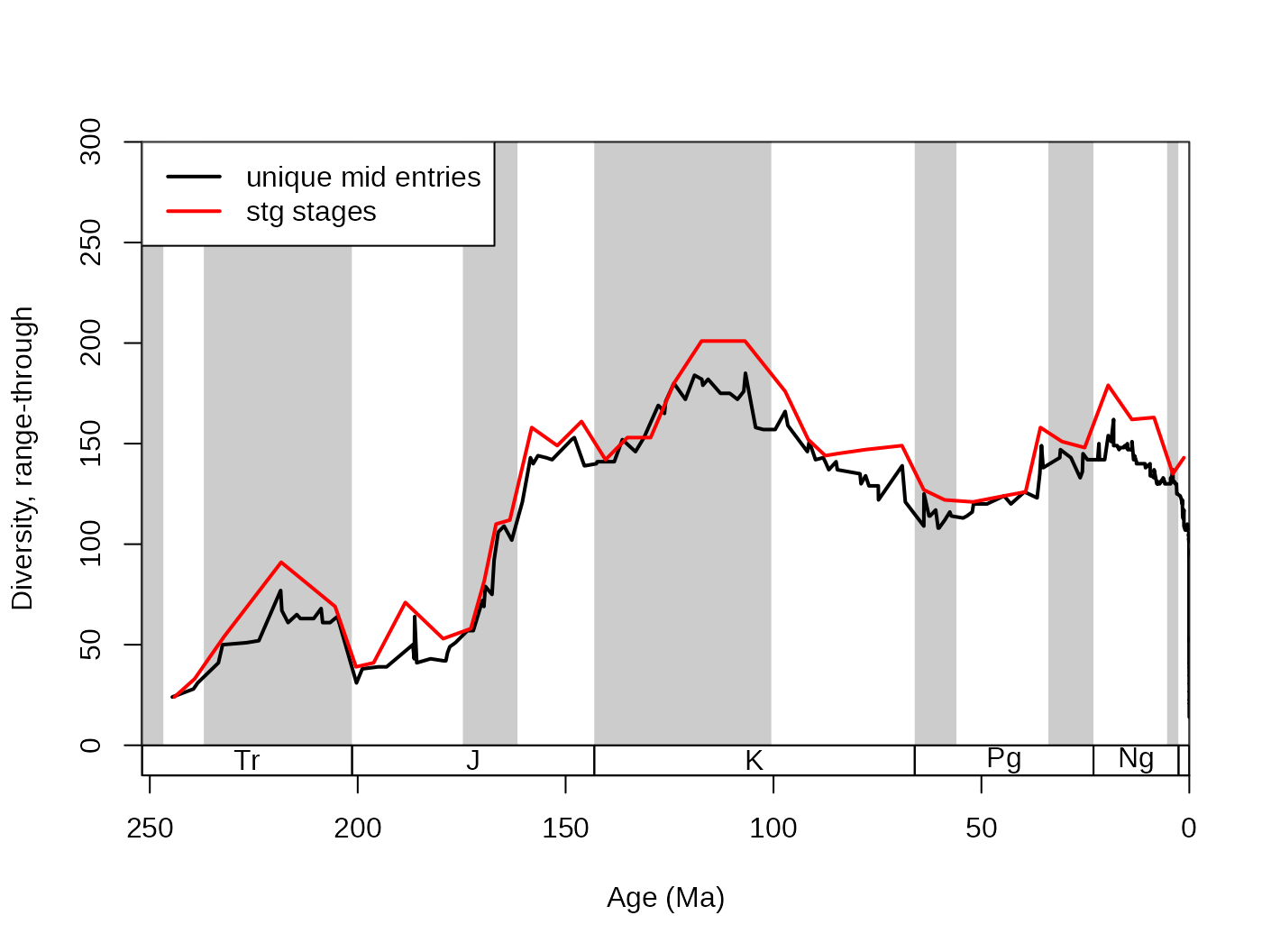
4.3.2. Slicing: slice() and the age
argument of divDyn()
The term slicing refers to the discretization of continuous time. A
potentially more promising solution is to assign the occurrences that go
along a continuous time scale to regularly spaced bins. However, we have
to keep the direction of time in mind: age values decrease as time
passes by. This is achieved with the slice() function, that
can also produce a time scale object, similarly to the
stages data.frame. This function converts a
continuous time vector to positive integers. To achieve this, you have
to supply a numeric vector to the breaks argument function.
The entries in this vector will represent the breakpoints or boundaries
of the interval, similarly to the breaks argument of the
cut() or hist() function in the base R
distribution. For the sake of simplicity, let’s try a 10 million year
bin resolution.
# resolve time
breakPoints <- seq(270, 0, -10)
sliTen <- slice(fossils$mid_ma, breaks=breakPoints, ts=TRUE)
str(sliTen)## List of 2
## $ slc: num [1:29544] 11 12 12 12 12 12 12 16 16 16 ...
## $ ts :'data.frame': 27 obs. of 4 variables:
## ..$ bottom: num [1:27] 270 260 250 240 230 220 210 200 190 180 ...
## ..$ mid : num [1:27] 265 255 245 235 225 215 205 195 185 175 ...
## ..$ top : num [1:27] 260 250 240 230 220 210 200 190 180 170 ...
## ..$ slc : num [1:27] 1 2 3 4 5 6 7 8 9 10 ...The first element of the output list ($slc) is the
discretized version of the original vector, with reversed time: in the
original vector it was flowing from higher to lower values, whereas in
$slc time flows from lower to higher values. The second
element is the time scale object that is synthesized from the given
breaks. It also has an $slc column that
reflects the connection between the age estimates and the slice numbers.
This output can be used to run the divDyn() function and to
visualize the results
# assign new column to the data frame
fossils$slc <- sliTen$slc
# run divDyn with the new column
auto10<-divDyn(fossils, tax="genus", bin="slc")
auto10$divRT## [1] NA NA 24 56 55 87 77 45 73 80 150 165 165 165 194 197 189 184 150
## [20] 156 165 126 124 173 165 183 177Note, that the rows of the divDyn() output are aligned with the rows
of the slice() output sliTen$ts. The results
are practically identical to when you do the binning automatically with
the age argument of the divDyn() function.
# and calculate diversity dynamics
ddMid10<-divDyn(fossils, tax="genus", age="mid_ma", breaks=breakPoints)
ddMid10$divRT## [1] 0 0 24 56 55 87 77 45 73 80 150 165 165 165 194 197 189 184 150
## [20] 156 165 126 124 173 165 183 177In this case, the age variable of the output will
contain the mid ages of the discrete intervals. Do not worry about
setting the exact values for the start and end of slicing
(270 and 0 in the example above). If no data
occurs in the outlined bins, then the corresponding rows will be empty
in the table.
# lines
lines(ddMid10$mid_ma, ddMid10$divRT, col="blue", lwd=2)
# new legend
legend("topleft", legend=c("age estimates, 10MY",
"stg stages", "unique mid entries"), col=c("blue", "red", "black"),
lwd=c(2,2,2), bg="white")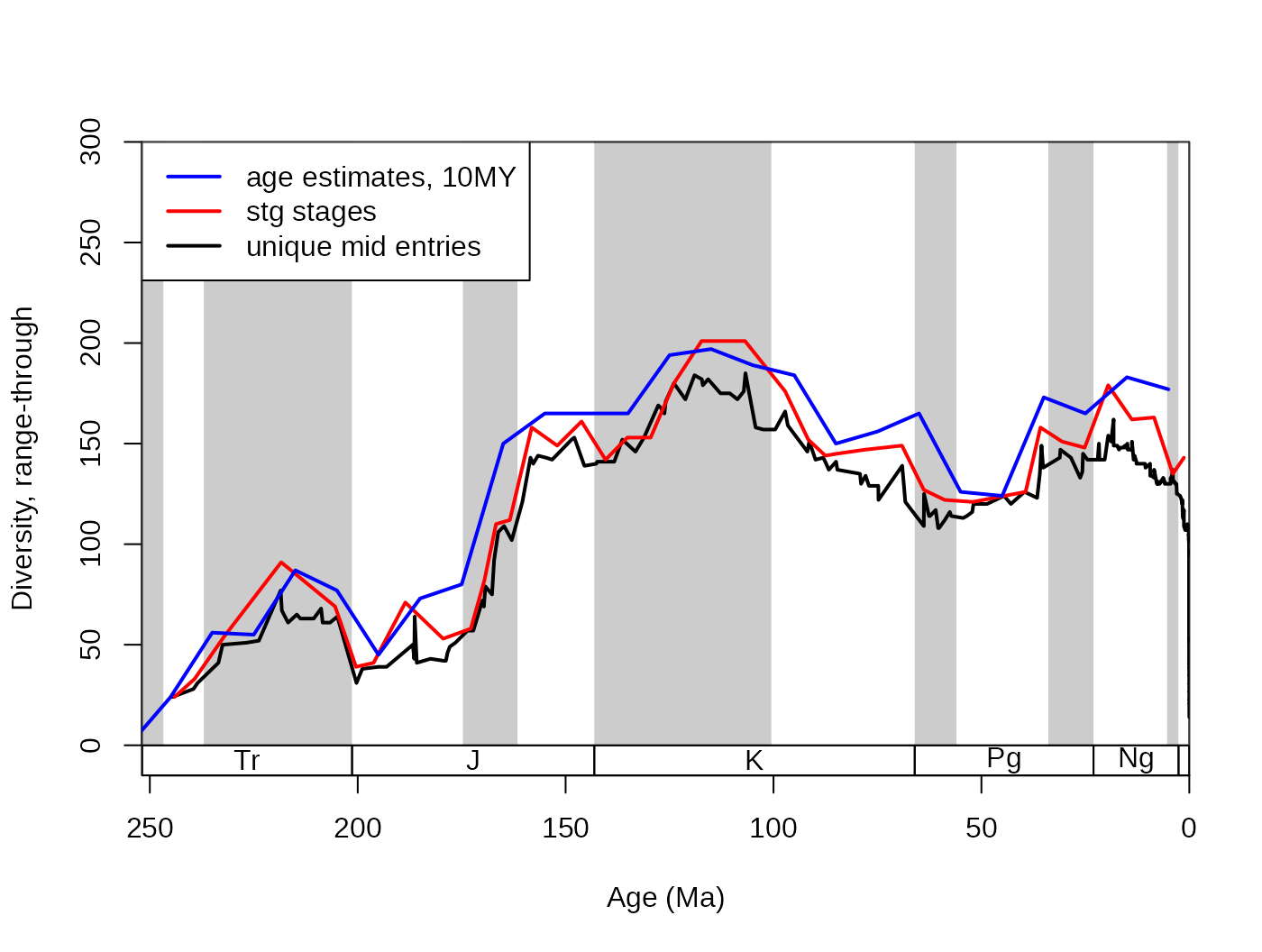
4.3.3. Summary – about slicing and the direction of time
It is better practice to use discretized time for analyses, as the
nature of the data supports it better. Interval names are more
conservative than age estimates that tend to vary with the evolving
geologic timescale, whereas the stratigraphic positions of fossils
barely change. In case you only have age estimates, I recommend to bin
them separately first with the slice() function, rather
than using the age argument of the divDyn()
function. The integer bins can be used in any procedures, such as
selectivity testing and subsampling, and you get better control over
your calculations.
When time is fed to the divDyn function with its
bin argument, it will assume by default that you are using
discrete bins, with identifiers that increase as time passes by. On the
other hand, using the age argument will assume that you use
ages, with identifiers that decrease with flow of time. As the
slice() function is designed to process age data, the way
it handles time is similar to the age argument of the
divDyn() function. You can override the default behavior in
all by setting revtime=TRUE. In short:
bin uses forward time, while age and
slice() use backward time.
4.4. Origination and extinction dates in binary response variables
The occurrence dataset can be represented a taxon/bin matrix that
taxa populate with presences or absences. Among the cells in this
matrix, some mark the apparent origination or extinction dates of taxa.
Two binary matrices can be outlined from this matrix: one for
extinctions, and one for originations. Cells, where the taxa are assumed
to be present are marked by 0s, and cells corresponding to the
origination (or extinction) dates of taxa contains 1s (the rest are
missing). Linearizing these matrices to binary vectors allows the
application of statistical modelling techniques to infer which entries
in additional variables are associated with the apparent origination and
extinction events. You can transform the occurrence dataset to a data
frame of such structure with the modeltab() function:
# basic function call
mtab <- modeltab(corals, tax="genus", bin="stg")
mtab[mtab[,"genus"]=="Acanthogyra",]## ori ext genus stg
## 8 TRUE FALSE Acanthogyra 67
## 9 FALSE FALSE Acanthogyra 68
## 10 FALSE FALSE Acanthogyra 69
## 11 FALSE FALSE Acanthogyra 74
## 12 FALSE TRUE Acanthogyra 75The ext variable stands for extinctions and the
ori column corresponds to originations. The basic output of
the function only includes rows corresponding to the time bin/taxon
matrix where the taxa are sampled. However, one could argue that
intervals where the taxa are assumed to be present (although they are
not sampled) are legitimate observations that they do not go extinct or
originate. You can add these interpolated points by setting the
rt argument of the function to TRUE:
# basic function call
mtabrt <- modeltab(corals, tax="genus", bin="stg", rt=TRUE)
mtabrt[mtabrt[,"genus"]=="Acanthogyra",]## occurrence ori ext genus stg
## 8 TRUE TRUE FALSE Acanthogyra 67
## 9 TRUE FALSE FALSE Acanthogyra 68
## 10 TRUE FALSE FALSE Acanthogyra 69
## 11 FALSE FALSE FALSE Acanthogyra 70
## 12 FALSE FALSE FALSE Acanthogyra 71
## 13 FALSE FALSE FALSE Acanthogyra 72
## 14 FALSE FALSE FALSE Acanthogyra 73
## 15 TRUE FALSE FALSE Acanthogyra 74
## 16 TRUE FALSE TRUE Acanthogyra 75Interpolated “occurrences” are marked with FALSE values
in the occurrence variable of the output dataset. It is
important to mention that this method is vulnerable to patterns of
incomplete sampling, as the rt extension of ranges only
uses interpolation, but no extrapolation.
Single-interval species require separate treatment as the
ext and ori response cannot be anything else
but TRUE. For this reason, these taxa are omitted by the
function by default (singletons=FALSE). If you want to
include these entries for some reason, you can do so by setting the
singletons argument to TRUE.
Additional variables can be directly added by specifying the original
column names of the taxon-specific entries (that have one to one
agreement with the taxon entries defined in the tax ) as
the taxvars argument of modeltab():
# function call with additional taxon-specific variables
modTab<- modeltab(corals, tax="genus", bin="stg",
rt=TRUE, taxvars=c("ecology", "growth"))
modTab[1:10,]## occurrence ori ext genus stg ecology growth
## 1 TRUE TRUE FALSE Acanthastrea 89 z colonial
## 2 TRUE FALSE FALSE Acanthastrea 90 z colonial
## 3 TRUE FALSE FALSE Acanthastrea 91 z colonial
## 4 TRUE FALSE FALSE Acanthastrea 92 z colonial
## 5 TRUE FALSE FALSE Acanthastrea 93 z colonial
## 6 TRUE FALSE FALSE Acanthastrea 94 z colonial
## 7 TRUE FALSE TRUE Acanthastrea 95 z colonial
## 8 TRUE TRUE FALSE Acanthogyra 67 z colonial
## 9 TRUE FALSE FALSE Acanthogyra 68 z colonial
## 10 TRUE FALSE FALSE Acanthogyra 69 z colonialYou are encouraged to use these structures as a framework for modelling the extinction and origination events by concatenating your own variables to this data.frame. Then you can use your preferred modelling technique, with calls such as:
simpleMod<- glm(ext ~ ecology + growth, family="binomial", data=modTab)5. Sampling standardization
5.1. Concepts
5.1.1. The goal of sampling standardization
Raw patterns of biodiversity are biased by heterogeneous, incomplete sampling. Consider the built-in example of the coral subset, where both the number of occurrences and the number of collections vary drastically over time.
sam <-binstat(fossils, bin="stg", tax="genus", coll="collection_no", duplicates=F)
cor.test(dd$divRT, sam$occs, method="spearman")## Warning in cor.test.default(dd$divRT, sam$occs, method = "spearman"): Cannot
## compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: dd$divRT and sam$occs
## S = 4708.8, p-value = 4.944e-05
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.589824
cor.test(dd$divSIB, sam$occs, method="spearman")## Warning in cor.test.default(dd$divSIB, sam$occs, method = "spearman"): Cannot
## compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: dd$divSIB and sam$occs
## S = 1453.9, p-value = 9.464e-14
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.8733499Sampling standardization allows the researcher to answer the question: would the patterns change, if sampling was homogeneous over time? There are two ways to answer this question: extrapolation and interpolation. Extrapolation methods try to infer on information we do not have. Interpolation methods, on the other hand, omit information to make sampling intensity comparable. The latter are more frequently employed in the paleontological context, and is usually referred to as ‘subsampling’, while the former has more robust applications when sampling is already quite extensive. Beware: as subsampling omits information, the result gained by subsampling is not an unequivocal substitute to the raw patterns, but rather their confirmation!
5.1.2. The formal representation of subsampling
Although rarefaction is defined as a deterministic estimator, due to the advancement of computational speed, recent implementations of the process use Monte Carlo estimation to get expected results of the subsampling procedure. This just means that a random subset of a given size is taken from the data, the desired statistic (e.g. taxonomic richness) is calculated, and these steps are iterated. The omission of information effectively simulates lower levels of sampling (but conditioned on the realized sampling).
This general question ‘what would the pattern would be like?’ is not specific to estimators of richness, but is generalizable to all other statistics that depend on sampling intensity. If the result of this estimator (Res) is dependent on the information from the time slice, it is essentially just a function f of the data (D) at hand with potential additional arguments argF:
\[Res= f(D, argF).\]
The same metric can be calculated by using the subsampled data in the time slice, but first we have to calculate this set with the subsampling procedure. This is just another function g that outputs a subset of the input dataset, to which we will refer to as the ‘trial dataset’:
\[sub = g(D, argD)\]
Therefore, the metric in question in a trial is dependent on both the two functions and their additional arguments. We can generalize such an implementation of subsampling with the following notation:
\[f(g(D, argG), argF)\]
resulting in the trial result. However this only applies to simple estimates that only use information from a single bin (e.g. simple diversity metrics such as SIB). But most other metrics are dependent on multiple slices, which means that the function result can be formalized as
\[res = f(D1, D2,... Dn, argF).\]
In order to make sampling standardization work, the subsampling procedure has to be applied to every bin-specific datasets. This means that a single trial result will be
\[tri = f(g(D1, argG), g(D2, argG),..., g(Di, argG), argF) \]
As the trial result is the manifestation of the randomness in
procedure g it has to be recalculated iteratively. In order to extract
the expectation of this procedure, the results will also have to be
averaged in some way, both of which are implemented in the
subsample() function. With this notation, the function f is
the applied function which can be specified with the
FUN argument, the g is the subsampler function,
which includes predefined procedures (will be expanded so that the users
can write custom procedures). The arguments argF and
argG are the applied and subsampling
arguments respectively. Naturally, you can use the function with
different instances that have the same abstract representation. The
basic arguments of subsampling are discussed in the next chapter.
5.2. Basic arguments
Some arguments must be provided in order to make the function run.
These will be demonstrated with the simplest, Classical Rarefaction
method (CR, Raup, 1975). All other subsampling methods (see section
‘5.4. Different Subsampling Types) use these input parameters, but most
of them will require additional arguments. In all cases, you will have
to specify the input dataset, the column containing the categories
(taxon names) and the binning variable, by which the data will be
dissected to run the subsampler function. These are the x,
tax, and bin arguments, respectively. As the
function is expected to be used on Paleobiology Database data, the
duplicates argument (see section below on this argument) is set to
FALSE by default for quicker use in these cases. This
necessitates the ‘coll’ argument that defines the column name of the
collection identifiers, which is set to the database default
collection_no. This variable designates samples in the
occurrence dataset. If you do not have such samples, then set the
duplicates argument to TRUE, which makes it possible to run the function
without such a variable.
5.2.1. Subsampling level
The only other mandatory input is the ‘level’ of subsampling, which
can be specified using the q argument. The exact nature of
this value is dependent on the subsampler function (see section
‘Different Subsampling Methods’), but in general it expresses sampling
intensity. For the CR subsampling method, this argument represents the
subsampling quota, the desired number of occurrences in the ‘trial
datasets’. Increasing values will result in increasingly higher sampled
diversities. The ‘q’ argument cannot be negative. With the subsampling
quota of 40 and 60 occurrences the function outputs the following
results for the coral dataset:
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="SIB diversity", ylim=c(0,200))
# raw diversity
lines(stages$mid[1:94], dd$divCSIB, col="black", lwd=2)
# subsampling
# with 40 occs.
subCR40 <-subsample(fossils, bin="stg", tax="genus", q=40)
lines(stages$mid[1:94], subCR40$divCSIB, col="blue", lwd=2)
# with 80 occs.
subCR80 <-subsample(fossils, bin="stg", tax="genus", q=80)
lines(stages$mid[1:94], subCR80$divCSIB, col="red", lwd=2)
# legend
legend("topleft", legend=c("raw", "CR, q = 40","CR, q = 80"),
col=c("black", "blue", "red"), lwd=c(2,2,2), bg="white")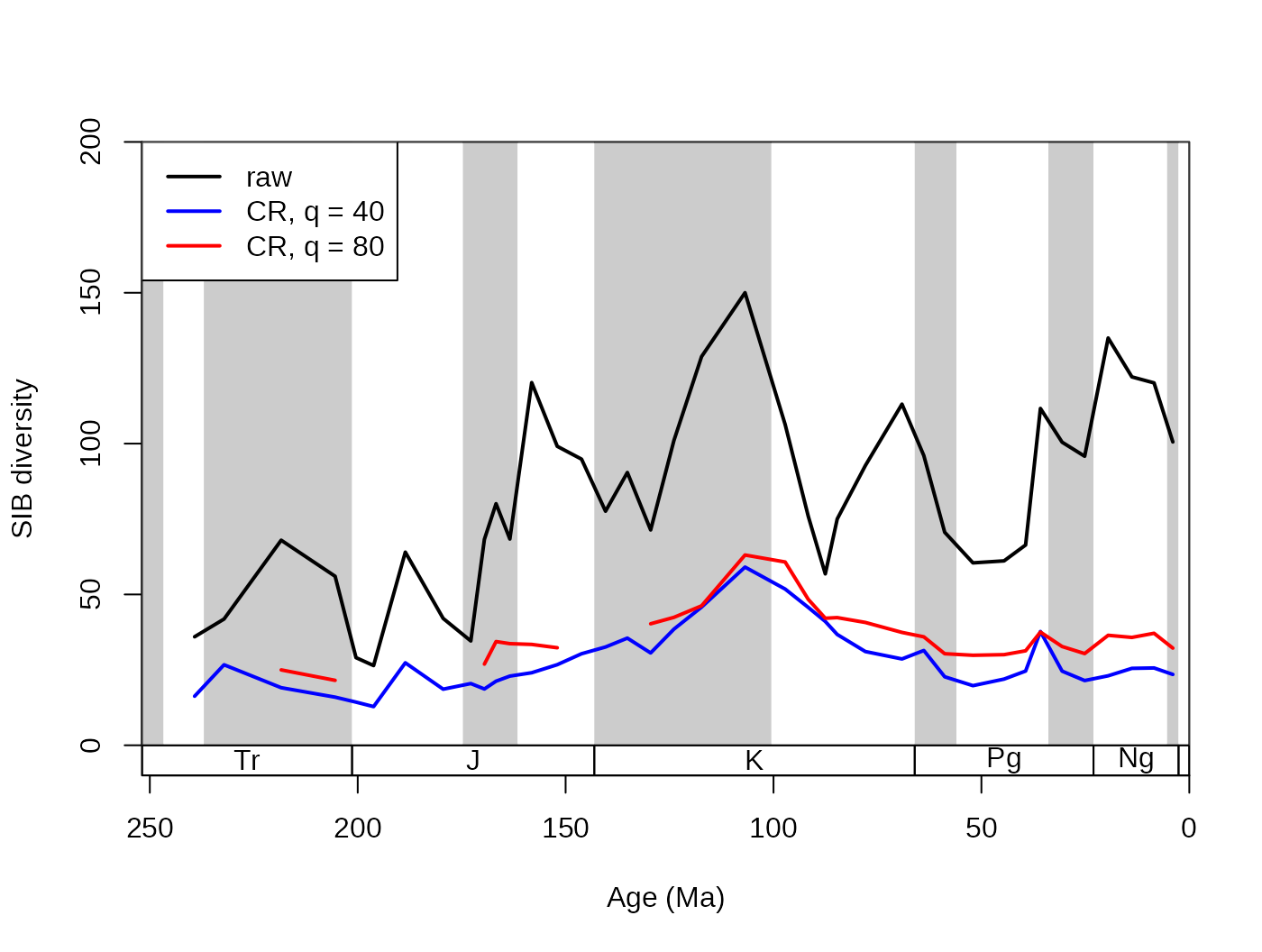
5.2.2. The number of iterations/trials
The precision of the simulations can be chosen by changing the number
of iterations the function will run. This can set with the
iter argument that has to be a single positive integer. In
general, the more iterations, the better the results, but the time the
function needs to run has a linear relationship with the iteration
number. Most paleontological examples are stable after a couple hundred
iterations. However, if the result of the function changes with every
run, you will have to increase this number!
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="genus richness (corrected SIB)", ylim=c(0,200))
lines(stages$mid[1:94], dd$divCSIB, col="black", lwd=2)
## subsampled, stable
subStab <-subsample(fossils, bin="stg", tax="genus", iter=100, q=30)
lines(stages$mid[1:94], subStab$divCSIB, col="blue", lwd=2)
## subsampled, unstable
subInstab <-subsample(fossils, bin="stg", tax="genus", iter=5, q=30)
lines(stages$mid[1:94], subInstab$divCSIB, col="red", lwd=2)
# legend
legend("topleft", legend=c("raw", "CR, q=30, stable",
"CR, q=30, unstable"), col=c("black", "blue", "red"),
lwd=c(2,2,2), bg="white")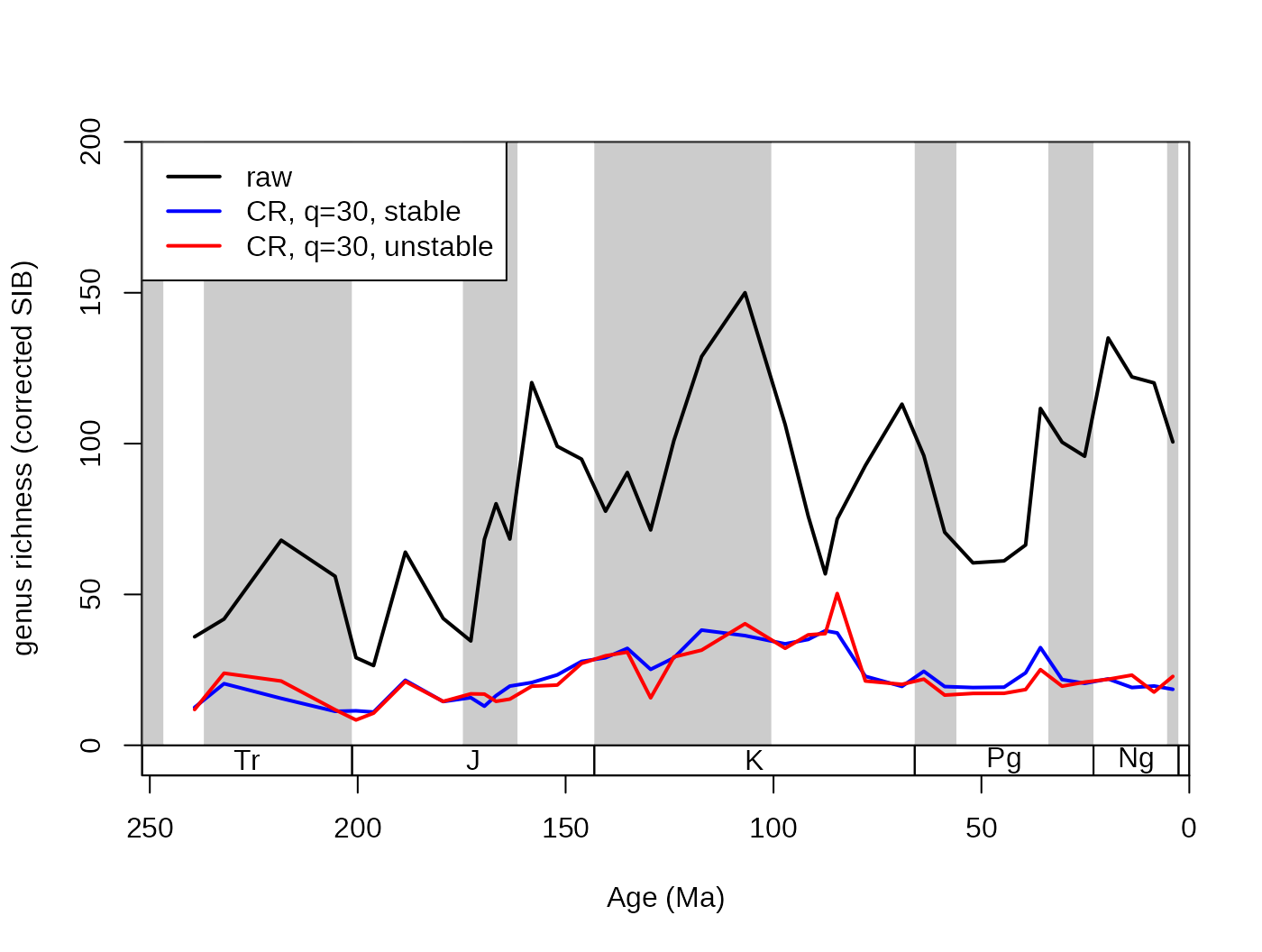
In this example, the red line is based on only 5 iterations, while the blue one is based on 100. This typically means that the low iteration number will have similar trajectory to the other series, but will have more volatility due to the random sampling error.
5.2.3. The duplicates argument
In the Paleobiology Database, occurrences are organized in
collections, and are recorded as lists of taxa. These entries optimally
are on the species-level of taxonomic resolution while most analyses
operate at the level of genera. As multiple species can be registered in
a single collection, the same genus name can appear multiple time in the
collection list. This will have an effect on the occurrence-based
subsampling methods. Using the duplicates arguments, you
can toggle whether the surplus entries should be omitted
(duplicates=FALSE, default) or whether they should be kept
(duplicates=TRUE). The omission is applied to the variables
defined in tax and coll variables. In case
duplicates=FALSE the coll argument is
mandatory.
## subsampled, stable
subCRnd <- subsample(fossils, bin="stg", tax="genus",
coll="collection_no", iter=100, q=40, duplicates=FALSE)
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="number of occurrences", ylim=c(0,100))
lines(stages$mid[1:94], subCR40$divCSIB, col="blue", lwd=2)
lines(stages$mid[1:94], subCRnd$divCSIB, col="black", lwd=2)
legend("topleft", legend=c("CR, q=40 (with duplicates)",
"CR, q=40"), col=c("blue", "black"),
lwd=c(2,2), bg="white")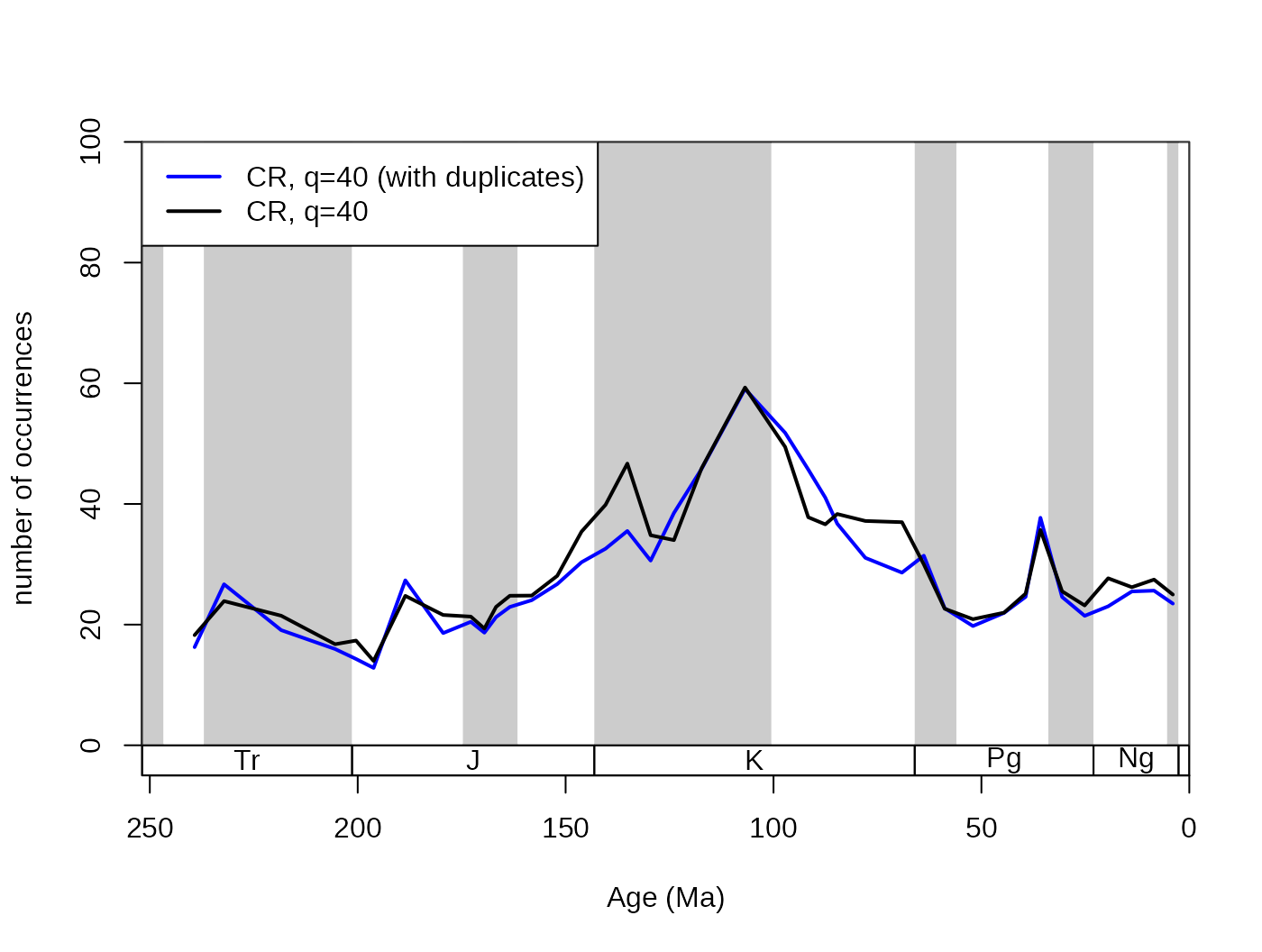 This particular dataset actually contains multiple genus occurrences, so
changing this argument to
This particular dataset actually contains multiple genus occurrences, so
changing this argument to FALSE is justified. However, as
it lengthens the function call considerably, the following examples will
be based on data that already had this filtering step:
# indicate identical collection/genus combinations
collGenus <- paste(fossils$collection_no, fossils$genus)
# omit the duplicates from the occurrence datasets
fossGen <- fossils[!duplicated(collGenus),]Just remember that this step can be skipped by adding
duplicates=FALSE to the subsample() function
call.
5.2.4. The useFailed argument
The subsampling level can be set as high as the user wants it to be.
Depending on whether the data in the time slices reach the subsampling
quota or not, the time slices can be included or excluded from the
results. If the useFailed argument is set to
FALSE, then the time slices that do not have enough
information to reach the subsampling quota will be omitted from the
resulting series. This is the default setting, and if the applied
function output is a scalar or a vector, then the corresponding
results will be omitted. If useFailed=TRUE then the bins
where the quota is not reached will be represented in the trial
dataset with all their sampled occurrences.
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="number of occurrences", ylim=c(0,100))
# subsampled, without failed
withoutFail<-subsample(fossGen, bin="stg", tax="genus",
iter=100, q=80, useFailed=FALSE)
lines(stages$mid[1:94], withoutFail$divCSIB, col="black", lwd=2)
# subsampled, with failed
withFail <-subsample(fossGen, bin="stg", tax="genus",
iter=100, q=80, useFailed=TRUE)
lines(stages$mid[1:94], withFail$divCSIB, col="blue", lwd=2)
legend("topleft", legend=c("CR, q=80, without failed",
"CR, q=100, with failed"), col=c("black", "blue"),
lwd=c(2,2), bg="white")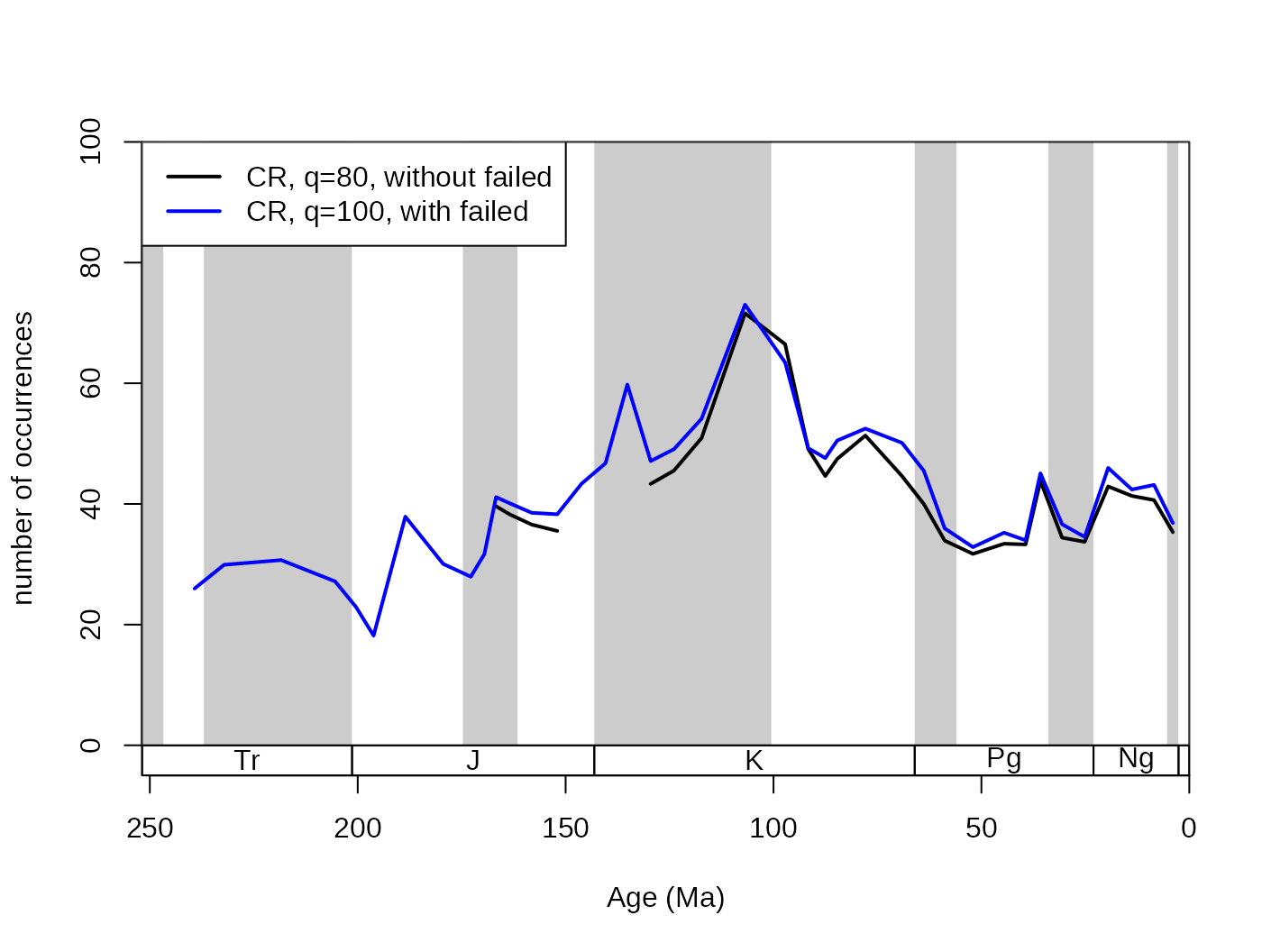
5.2.5. The keep and rem arguments
These arguments handle which bins should be forced to be kept
(included) or removed (excluded) from the trial dataset. It
accepts a numeric vector, with the bin identifiers. Positive entries
mean that the bins will be included in the trial dataset
without subsampling. This can be useful if you want the inclusion of
recent ‘occurrences’ to demonstrate the ‘Pull of the Recent’ effect.
This example uses the original corals dataset to
demonstrate how the argument works (with duplicates omitted).
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="per capita extinction rates", ylim=c(0,1))
## subsampled, excluding the recent occurrences
sub <- subsample(corals, bin="stg", tax="genus", iter=100,
q=50, rem= 95, duplicates=FALSE, coll="collection_no")
lines(stages$mid, sub$extPC, col="black", lwd=2)
## subsampled, including the recent
subPR <- subsample(corals, bin="stg", tax="genus", iter=100,
q=50, keep= 95, duplicates=FALSE, coll="collection_no")
lines(stages$mid, subPR$extPC, col="blue", lwd=2)
# legend
legend("topleft", legend=c("only fossils",
"including the recent\n(not subsampled)"), col=c("black", "blue"),
lwd=c(2,2), bg="white")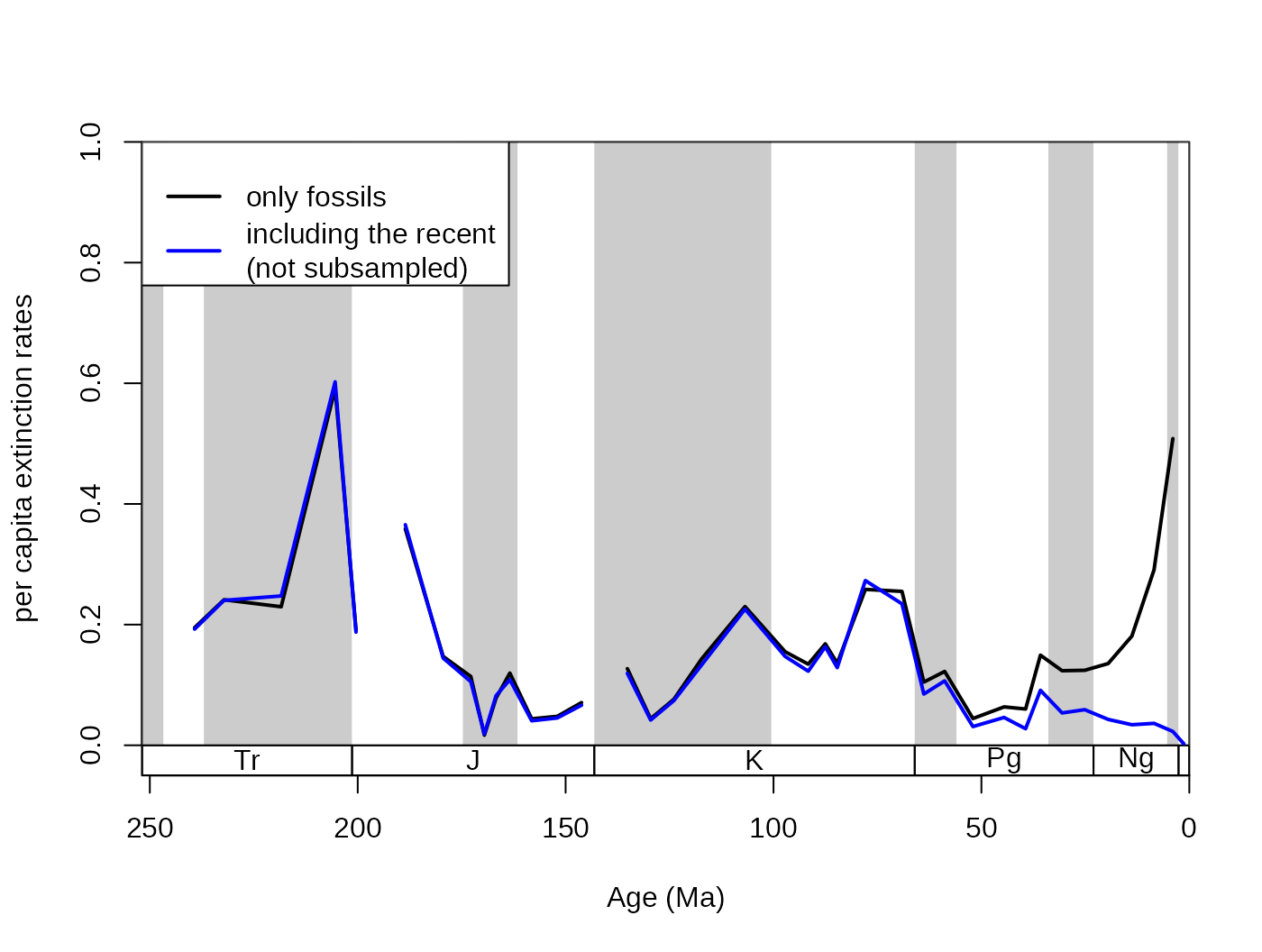
Between these two curves, neither shows perfectly accurate results.
The per capita rates employ the range-through assumption. The first
results (sub) is biased by edge effects, the closer the end
of the time series is, the higher the rate values are due to decreasing
total amount of range extensions. The inclusion of entries in the second
dataset on the other hand allows the ‘Pull of the Recent’ to depress the
extinction rates of the Late Cenozoic.
5.2.6. Subsampling ‘output’ and plotting options
The output type of the subsampling process can be set with the output argument. This should be dependent on the further use of the function results. The most direct output is the arithmetic (default) or geometric means of the trials. They provide almost the same output:
# basic plot
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="richness of genera (corrected SIB)", ylim=c(0,100))
## arithmetic mean output
subArit <- subsample(fossGen, bin="stg", tax="genus", iter=100, q=40, output="arit")
lines(stages$mid[1:94], subArit$divCSIB, col="black", lwd=2)
## geometric mean output
subGeom <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=40, output="geom")
lines(stages$mid[1:94], subGeom$divCSIB, col="blue", lwd=2)
legend("topleft", legend=c("arithmetic means", "geometric means"),
col=c("black", "blue"), lwd=c(2,2), bg="white")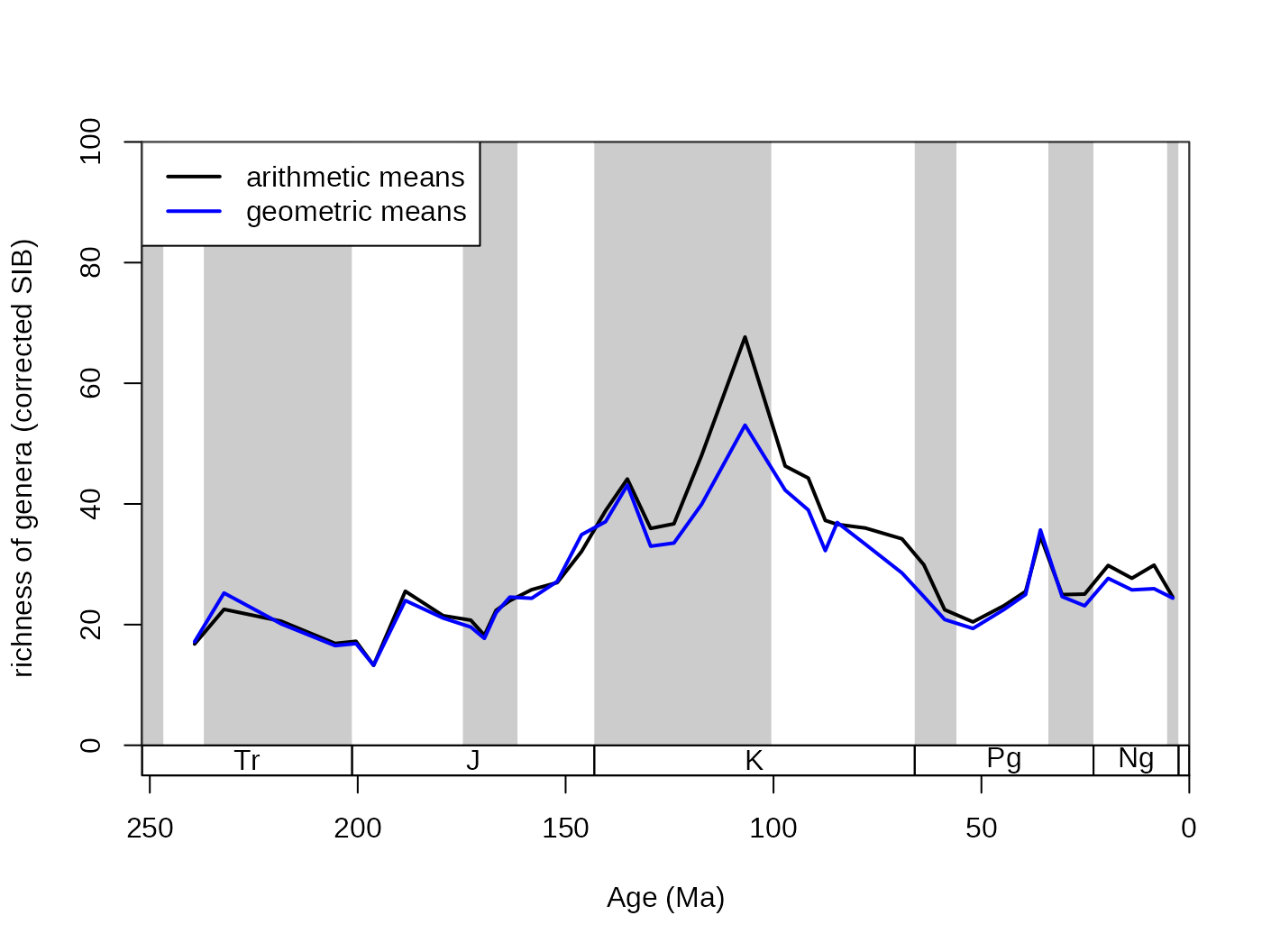
However, the subsampling outputs demonstrate the natural variance
arising from the simulating nature of the process. The results of the
individual trials can be conserved by setting the output argument to
either "dist" or "list". The difference
between these two options is that dist groups the results
of the trials by the structure of the original function. In the cases
when the divDyn() function is the applied function
(every example until now), the output is a list, where all variables are
matrices.
## subsampled, dist output
subDist <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=40, output="dist")Rows are time slices, while columns represent the individual trials.
These can be visualized by a simple for() loop, and the
arithmetic means can be calculated the regular way.
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="subsampled richness of genera (corrected SIB)", ylim=c(0,150))
plottedVar <- subDist$divCSIB
for(i in 1:ncol(plottedVar)){
lines(stages$mid[1:94], plottedVar[,i], col="#00000099", lwd=2)
}
# the mean
csibMeans<- apply(plottedVar,1, mean, na.rm=T)
lines(stages$mid[1:94], csibMeans , col="red", lwd=2)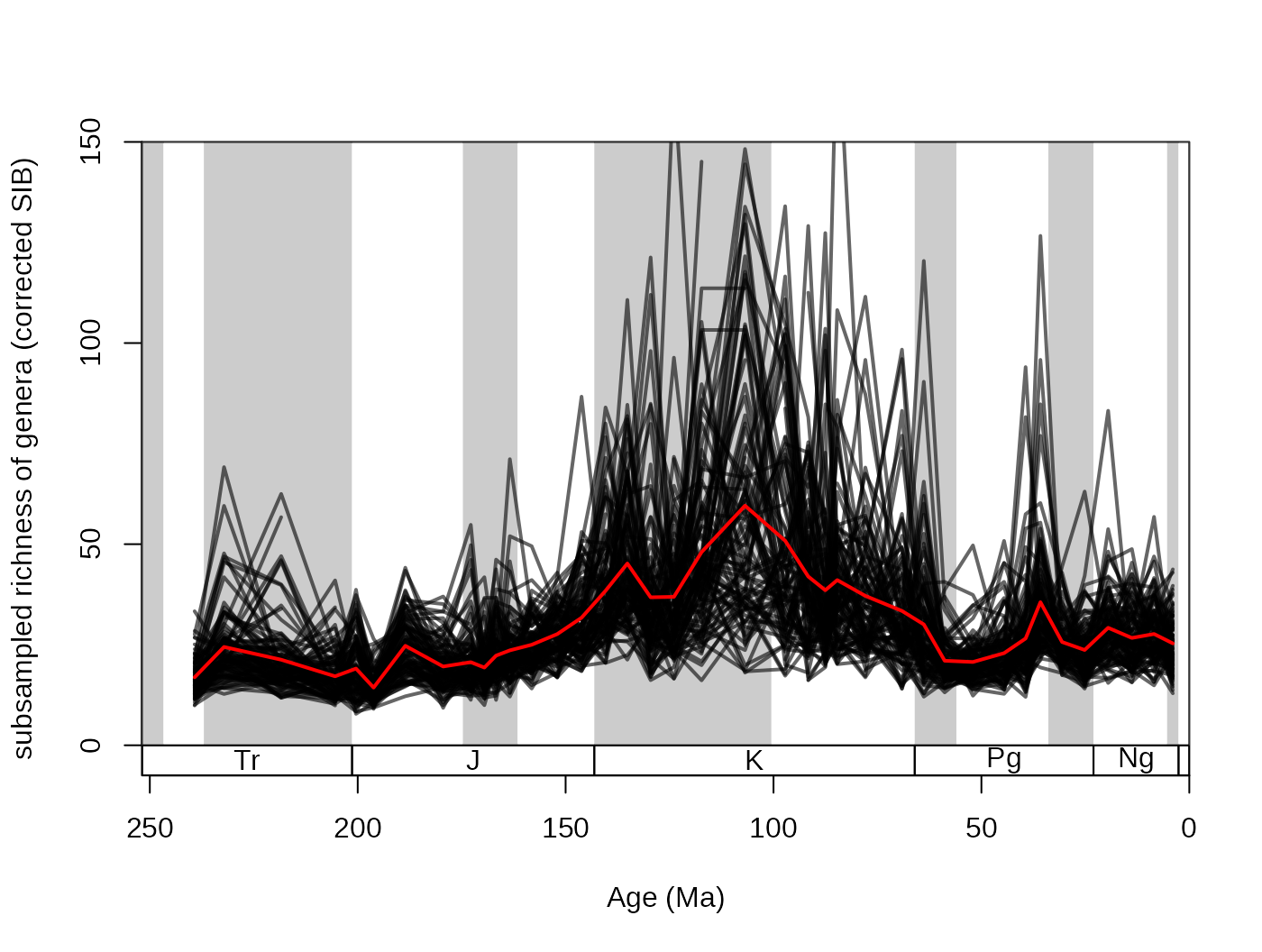
For convenience, this variance can also be plotted with the new
shades() function. That will display the distribution of
values by drawing transparent polygons. The quantiles of each
distribution are calculated, and the same values are then connected (.75
to .75).
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="subsampled richness of genera (corrected SIB)", ylim=c(0,150))
shades(stages$mid[1:94], plottedVar, res=10, col="black")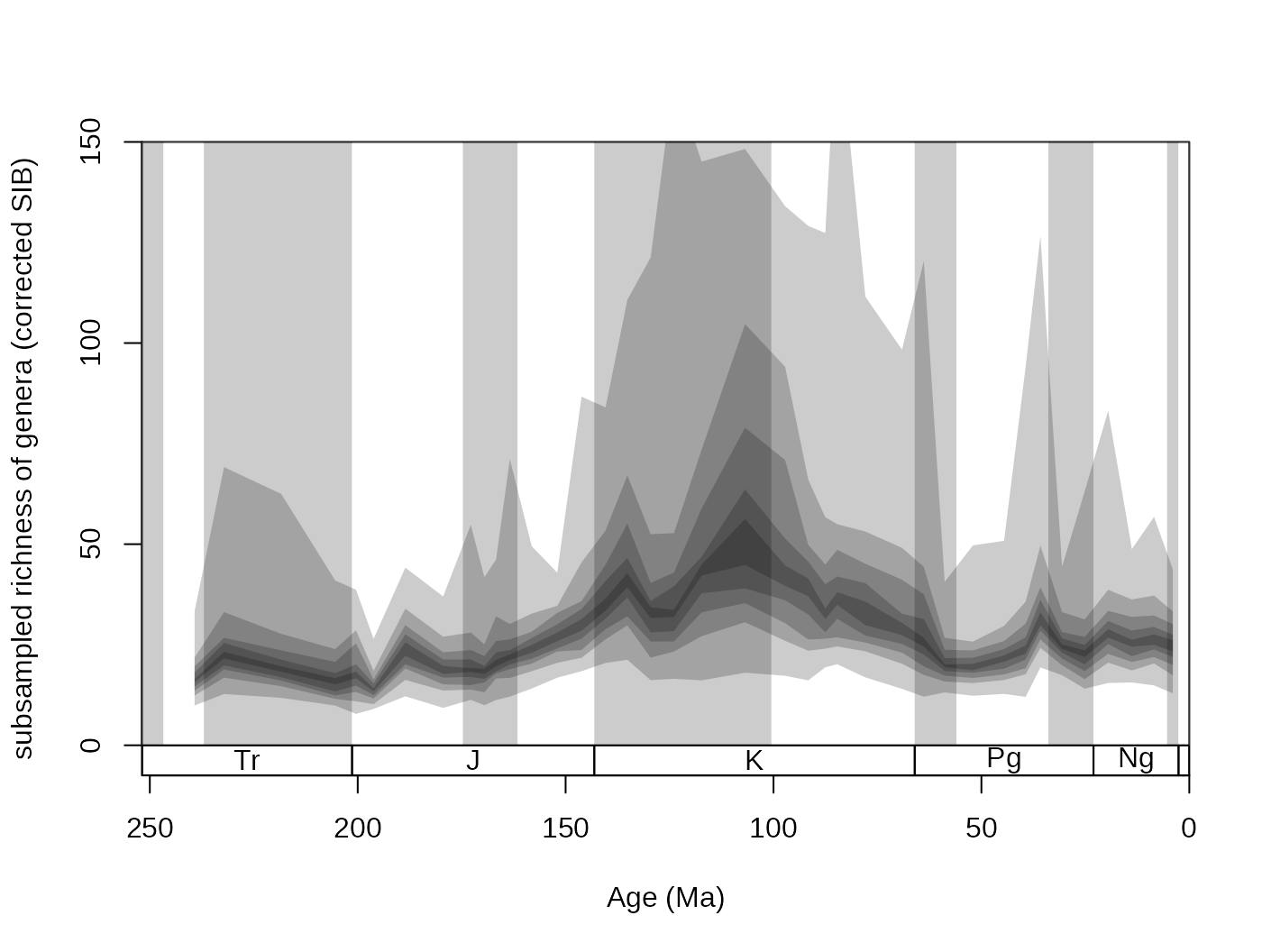
Setting the res argument of this function controls the
‘quantile resolution’. The higher the number, the more refined the
transparency gradient will be. However, it is more useful to the enter
the exact quantiles to be plotted in the form of a numeric vector:
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="subsampled richness of genera (corrected SIB)", ylim=c(0,150))
shades(stages$mid[1:94], plottedVar, col="blue", res=c(0.05,0.25,0.75,0.95))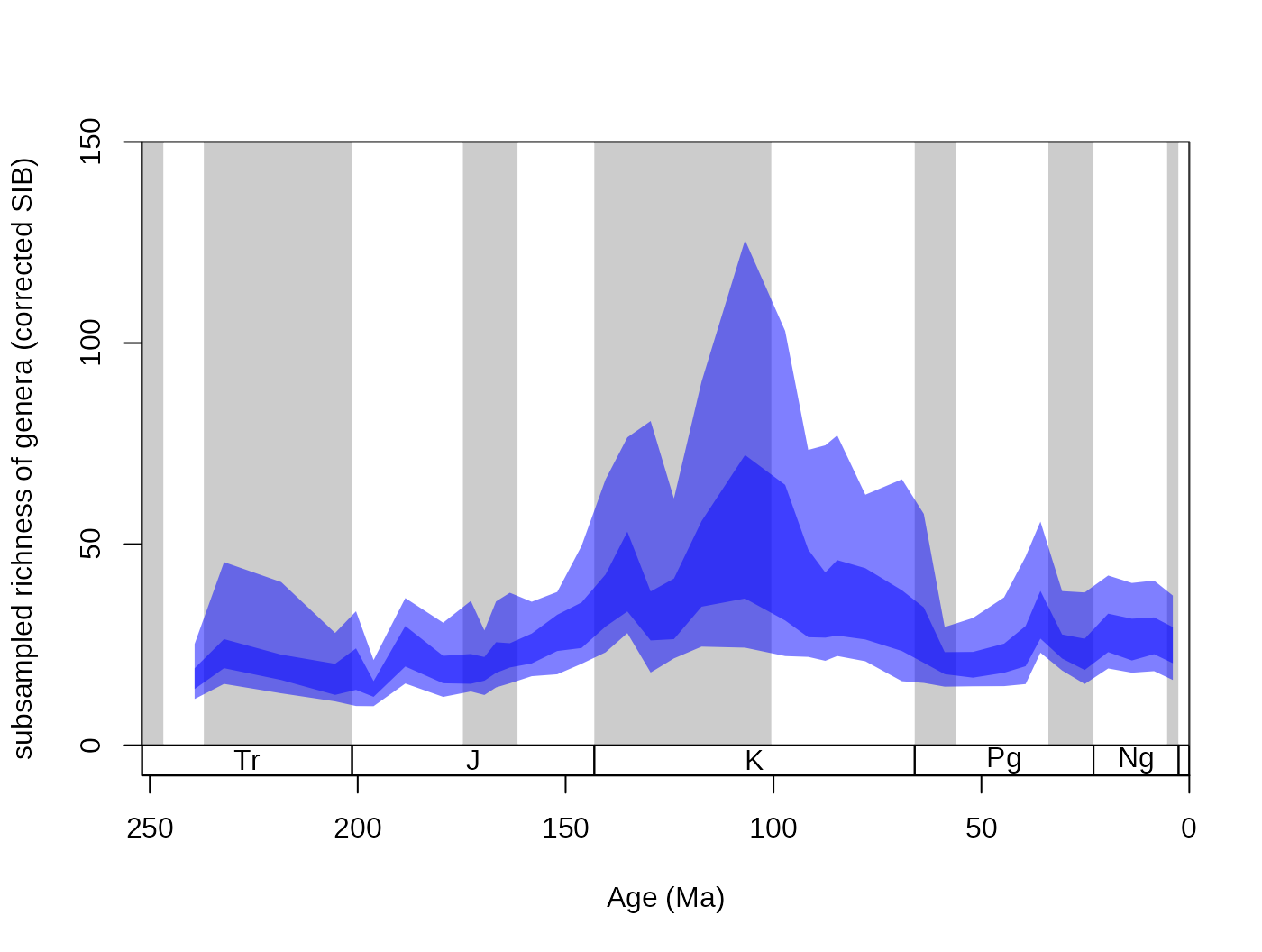
The function’s color argument only accepts colors that are specified
without the alpha channel (for instance "#00000066" is
impossible, but "blue" or "#4433FF" are
perfectly viable).
5.3. The applied function during the subsampling
By default, executing the subsample() command on the
occurrence data will run the divDyn() function in every
iteration, which is specified by the FUN argument (function
f in the notation above). Setting this argument to
NULL will not run any function on the subsampled data
subset of the trials (trial data). After all iterations are
finished, the data subsets will be concatenated and the function will
output them as a list of length iter. This option coerces
the output type of the function to list, no other output
type is possible.
## subsampled
subData <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=40,FUN= NULL)
# characteristics
class(subData)## [1] "list"
names(subData)## [1] "results" "failed"
length(subData$results)## [1] 100
# columns of the trial dataset
colnames(subData$results[[1]])## [1] "genus" "collection_no" "family" "abund_value"
## [5] "abund_unit" "reference_no" "life_habit" "diet"
## [9] "country" "geoplate" "lat" "lng"
## [13] "paleolat" "paleolng" "period" "epoch"
## [17] "subepoch" "stage" "early_interval" "late_interval"
## [21] "max_ma" "min_ma" "stg" "ten"
## [25] "env" "lith" "latgroup" "bath"
## [29] "gensp" "ecology" "ecologyMostZ" "ecologyMostAZ"
## [33] "ecologyBoth" "growth" "integration" "mid"
## [37] "stgMid" "mid_ma" "slc"The advantage of this option is that you can inspect the results of
the subsampling output. You can also iterate a custom function on this
output by using the lapply() interator in base R. The
results of the function will be output as a list.
5.3.1. Example 1: Checking the number of occurrences
One of the functions we can check is the number of occurrences in the
‘trial datasets’. By the rules of CR, this should be exactly
q:
OCC <- function(x) table(x$stg)
# list of trials, each contains the number of occurrences in a bin (vector)
subOccs <- lapply(subData$results, OCC)
# one trial
subOccs [[1]]##
## 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79
## 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40
## 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94
## 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40Extracting the central tendency (average of the trials) is more
difficult this way, but some output structures (e.g. geographic shapes)
might require special treatment that can be written as a custom
function. Anyway, subsample() allows the simplification of
this process, if you set the output argument to ‘list’ and by providing
the custom function as the FUN argument. One rule is that
the function must take the occurrence dataset as an argument, which is
formally called x:
OCC <- function(x) table(x$stg)
subOccsInternal <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=40,FUN= OCC, output="list")
subOccsInternal$results[[1]]##
## 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79
## 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40
## 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94
## 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40Whether averaging is possible will be dependent on the output of the
applied function. If the output of the vector is scalar, then
the output methods "arit" and "geom" will
return a scalar, and "dist" will return a vector. If the
output of the function is a vector, both "arit" and
"geom" will return a single vector, and "dist"
will return a matrix. If the result of the applied function is
a data.frame, then the result of "arit" and
"geom" will be a data.frame (from the result)
too. In this case, if the output is "dist", then the output
of the subsample function will be a list, each of its
elements representing one of the variables in the output of the
applied function, but instead of containing vectors, they will
contain matrices. This is the case for the divDyn()
function. Running the function OCC() above with the
"dist" type output will produce a matrix of
occurrences:
subDistOccs <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=40,FUN= OCC, output="dist")
str(subDistOccs)## int [1:41, 1:100] 40 40 40 40 40 40 40 40 40 40 ...
## - attr(*, "dimnames")=List of 2
## ..$ : chr [1:41] "54" "55" "56" "57" ...
## ..$ : NULLNote the dimensions of the matrix, which is 41 times
100, the number of sampled time slices and the number of
subsampling trials. CAUTION: The averaging and the grouping of the
resulting variables is possible because the applied function is
run on the total dataset first, which will result in a container
prototype that is used to store the trial results. If the
function output structure (i.e. dimensions) is different when it is run
for the subsets than when it is run on the total dataset, the subsample
function will output an error.
5.3.2. Example 2: maximum absolute paleolatitudes
Let’s say you are interested in the maximum absolute paleolatitude of the occurrences. This will be influenced by the number of occurrences, and should therefore be rechecked with subsampling. You can calculate this in the raw dataset with the following function, using the known variable names:
PL <- function(x){
tRes<- tapply(INDEX=x$stg, X=x$paleolat, FUN=function(y){
max(abs(y), na.rm=T)
})
return(tRes)
}
maxPaLat<- PL(fossils)This variable certainly increases with age,
cor.test(maxPaLat, stages$mid[54:94], method="spearman")##
## Spearman's rank correlation rho
##
## data: maxPaLat and stages$mid[54:94]
## S = 18336, p-value = 5.307e-05
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## -0.5972125but it is questionable at this point, whether the increasing number of occurrences is responsible for this pattern, or whether you would still see it, if the same number of occurrences were sampled from each time slice. You can quickly check this association at 20 occurrences and CR by running:
subMaxPaLat <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=20,FUN=PL)
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="maximum sampled paleolatitude", ylim=c(0,90))
lines(stages$mid[54:94], maxPaLat, col="blue")
lines(stages$mid[54:94], subMaxPaLat, col="black")
# legend
legend("topleft", legend=c("max",
"subsampled max"), col=c("blue","black"),
bg="white", lty=c(1,1))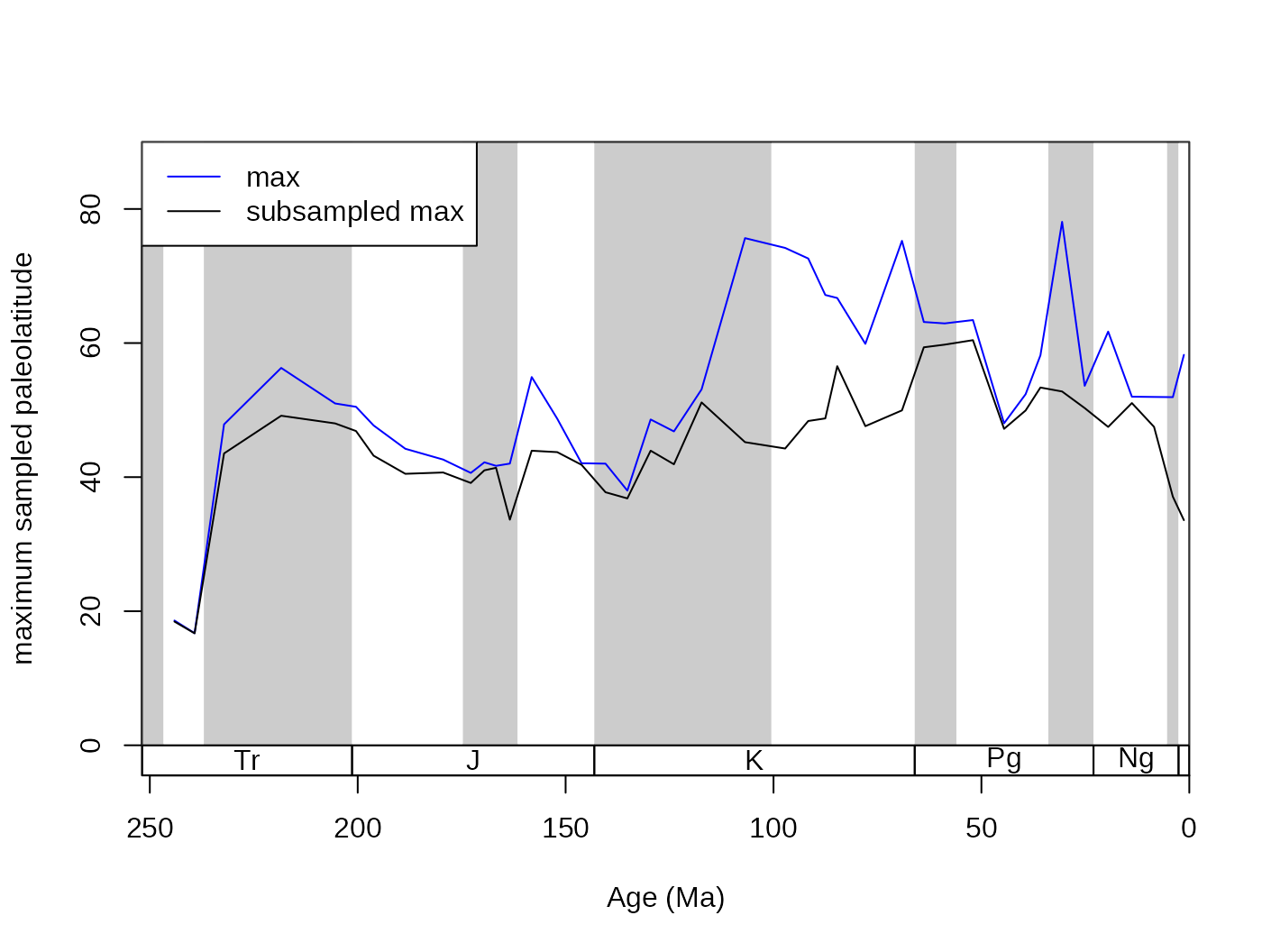
cor.test(subMaxPaLat, stages$mid[54:94], method="spearman")5.4. Different subsampling types
5.4.1. Classical Rarefaction (CR)
Classical Rarefaction is the most straightforward subsampling method.
It is based on the assumption that the number of occurrences is a direct
proxy for sampling intensity. Although this assumption can be criticized
(see below), the general applicability of the method, and
straightforward interpretation of the results makes it especially useful
for checking the distorting effects of sampling. The arguments of the
subsampling types are summarized at the help page of the subsampling
trial functions (5.4.4.). The traditional classical rarefaction
procedure was expanded to perform unit-based subsampling. In all cases
above, CR used the number of rows within a bin as the units of the
subsampling procedure which is set by the default argument
unit=NULL. However, the calculation can also be run with
multiple rows describing one unit (list) of the subsampling that is
indicated by another variable. This way you can rarefy the data to a
certain number of collections (UW subsampling, see below), references or
whatever, while the all rows forming the units remain intact.
subUWunit <- subsample(fossGen, bin="stg", tax="genus",
iter=100, q=10, type="cr", unit="collection_no")5.4.2. Occurrence-weighted by-list subsampling (OxW)
By-list subsampling methods use information about how the occurrences
are clustered in lists. In the PaleoDB, these clusters are the
collections that ‘contain’ the occurrences, which are direct products of
sampling. Depending on whether we would like to emphasize the sampling
of collections (related to beta diversity) or the number of entries in
lists, you can use the OxW group of subsampling (Alroy et
al., 2001). Since the development of SQS (or CBR, see below) these
methods have not been widely applied, but nevertheless, they could be
useful in some projects. The basis of these methods is that collection
integrity cannot be broken during the subsampling procedure. Entire
lists are drawn from the subsampling pool of each time bin, while the
number of occurrences are tracked. These lists are the collections that
should be indicated by setting the coll variable
appropriately. When the quota is reached, no more occurrences are drawn.
The rest of the process (assembly of the trial dataset, running
the applied function) is the same as with the CR method.
subOW <- subsample(fossGen, bin="stg", tax="genus", coll="collection_no",
iter=100, q=40, type="oxw")The output of the occurrence-weighted (OW) type is typically very close to the output of the corresponding level CR results:
subCR <- subsample(fossGen, bin="stg", tax="genus", iter=100, q=40)
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB diversity", ylim=c(0,90))
lines(stages$mid[1:94], subCR$divCSIB, col="black")
lines(stages$mid[1:94], subOW$divCSIB, col="blue")
legend("topleft", legend=c("CR - 40", "OW - 40"),
col=c("black", "blue"), lwd=c(2,2), bg="white")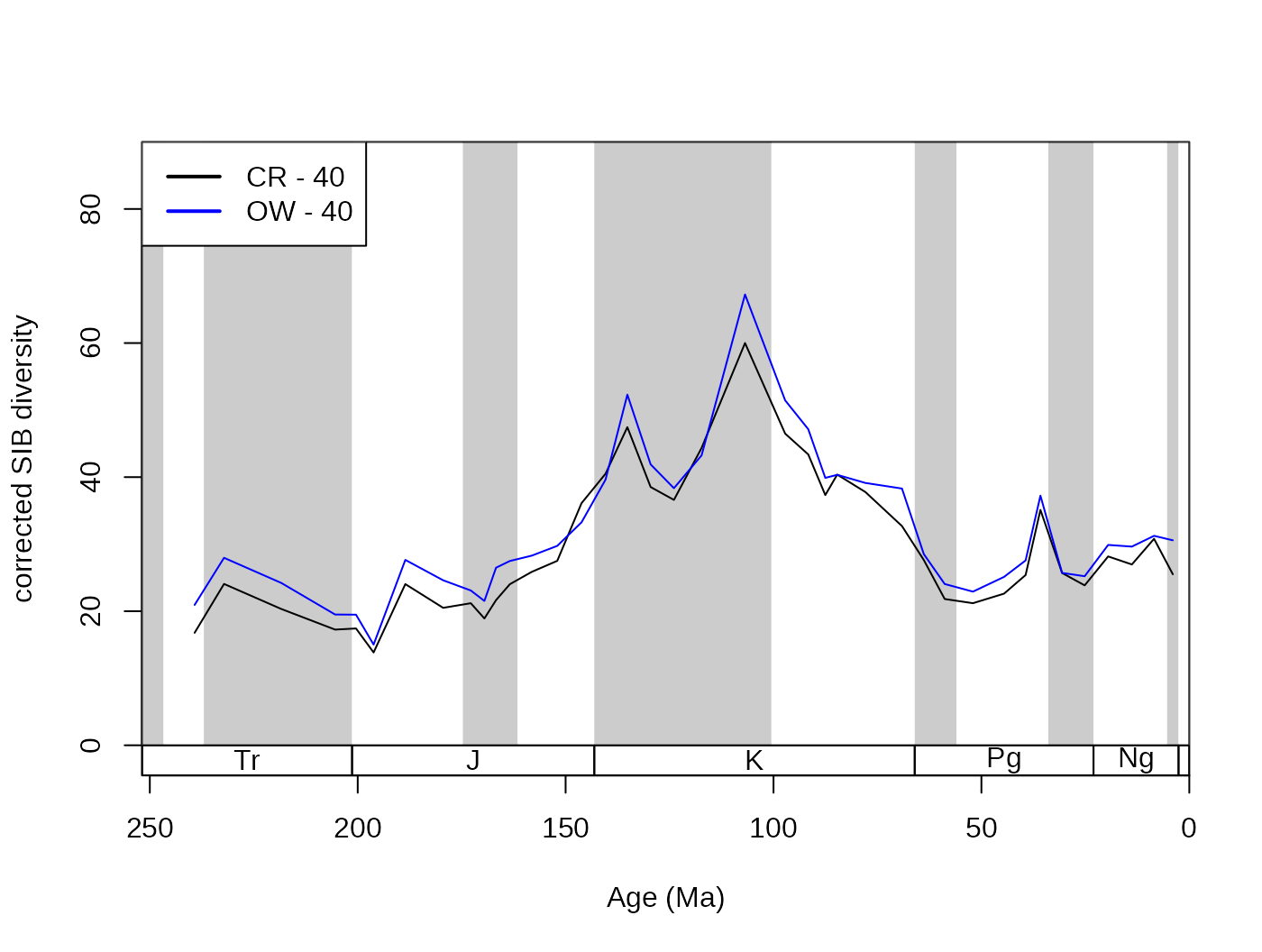
The reason why the method type is called the OxW is
because, depending on the relative importance of the collections and the
entries within the collection, lists express different aspects of
sampling. This can be taken into consideration with the exponentiation
of the occurrence counts in each collection. In these procedures, the
number of occurrences in each collection will be raised to the power of
xexp and the sum of the resulting values will be compared
to the set subsampling quota. The simplest way of doing this is to raise
the number of occurrences in a collection to the power of 0, which
effectively means the selection of a certain number of collections in
all different time slices (no matter how many occurrences there are in a
collection it will be 1 if raised to the power of 0). Setting the
xexp argument to 0, will force this setting. This is
sometimes referred to as the ‘unweighted subsampling’ method, which
effectively means to subsample the data to a certain number of
collections in a bin. In this case the q argument will
represent the quota of collections.
trialsUW <- subsample(fossGen, bin="stg", tax="genus", coll="collection_no",
iter=100, q=20, type="oxw", xexp=0, FUN=binstat)
# the number of sampled collections on average in each timeslice
trialsUW[54:94, "colls"]
subUW <- subsample(fossGen, bin="stg", tax="genus", coll="collection_no",
iter=100, q=10, type="oxw", xexp=0)By this principle, it is not difficult to see that depending on the
size of the collections, the actual number of occurrences will be
somewhat higher than the quota. You can quickly check the exact number
of occurrences drawn with the binstat() function
subST <- subsample(fossGen, bin="stg", tax="genus", coll="collection_no",
iter=100, q=20,FUN=binstat, type="oxw")
subST[54:94, "occs"]## [1] 22.93 22.80 24.50 24.41 24.99 23.54 21.82 23.88 21.93 23.97 22.65 25.76
## [13] 23.18 26.23 27.72 30.62 24.05 26.03 26.25 27.61 27.36 26.90 27.65 23.62
## [25] 26.99 25.71 24.98 24.66 22.89 24.36 22.36 23.43 23.06 27.17 26.13 26.13
## [37] 27.45 24.21 23.95 24.66 24.54Although there are ways to improve the accuracy of the subsampling
process, the variation induced by this problem is only minuscule
compared to those introduced by sampling and binning uncertainties.
However, typically this xexp value is set to a value
between 1 and 2 (e.g. 1.4). In these cases, you have to calculate the
intensity of sampling . Although there is some discrepancy between the
methods, the overall trajectories are quite similar.
subO2W <- subsample(fossGen, bin="stg", tax="genus", coll="collection_no",
iter=100, q=80, type="oxw", xexp=2)
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB genus richness", ylim=c(0,90))
lines(stages$mid[1:94], subCR$divCSIB, col="black")
lines(stages$mid[1:94], subOW$divCSIB, col="blue")
lines(stages$mid[1:94], subUW$divCSIB, col="red")
lines(stages$mid[1:94], subO2W$divCSIB, col="green")
legend("topleft", legend=c("CR - 40", "OW - 40", "UW - 10", "O2W - 80"),
col=c("black", "blue", "red", "green"), lwd=c(2,2), bg="white")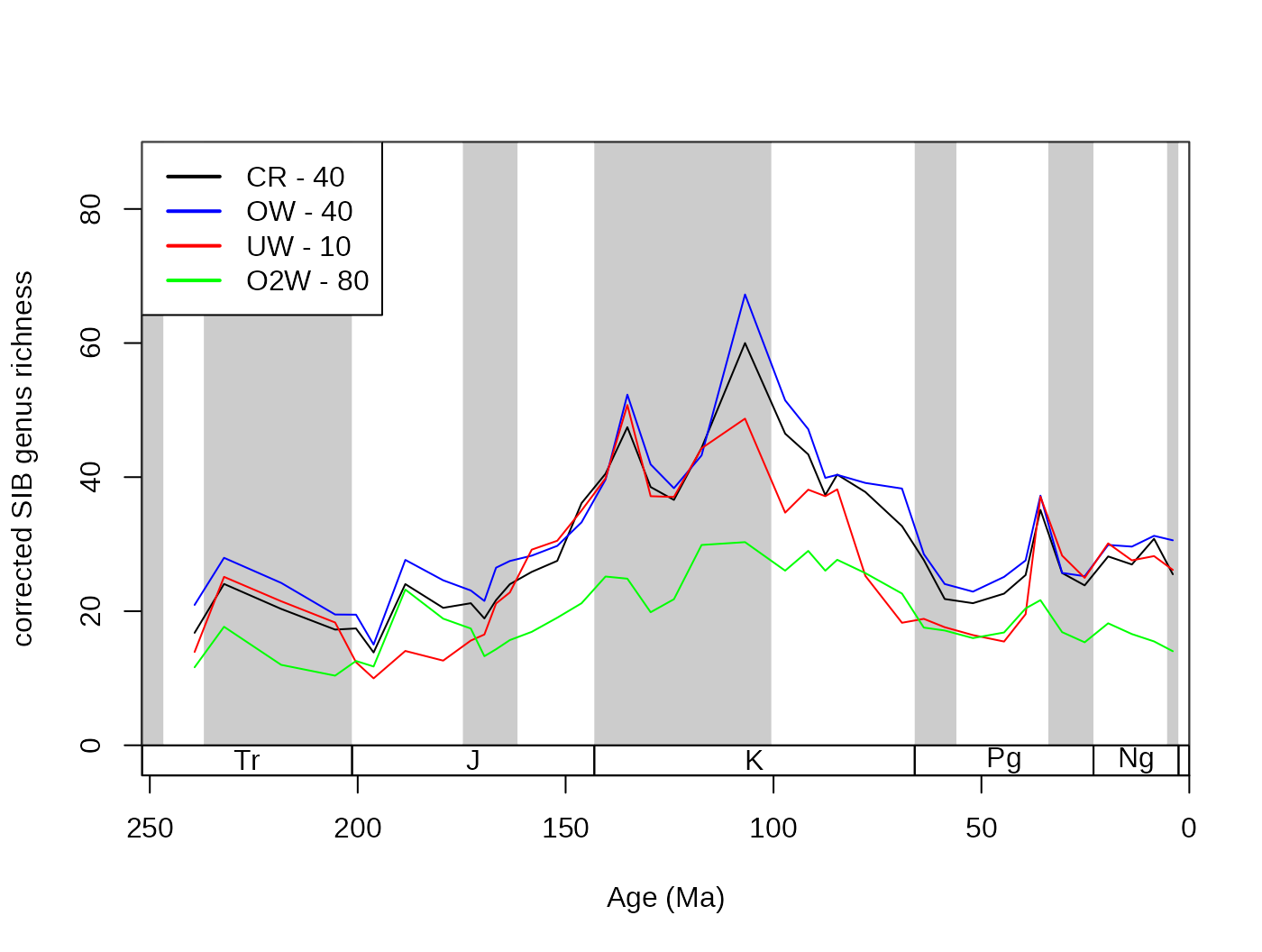
5.4.3. Shareholder Quorum Subsampling (SQS)
As intuitive as the CR approach is to understand, the method has been criticized for being ‘unfair’ (Alroy, 2010). The results of CR will not reflect changes of richness appropriately, if two localities with different diversities are compared and their total richnesses are not equal. This can be visualized by progressively lowering the subsampling quota for CR, which will result in not just decreased levels of richness but also in a pronounced decrease in variance (Alroy, 2010b). This effect can be illustrated by rerunning CR iteratively with progressively lower quotas.
# repeat CR subsampling for a set of quotas
quotas<-seq(70,10,-10)
for(i in quotas){
# actual CR
cr<-subsample(fossGen, iter=50, q=i,tax="genus", bin="stg",
useFailed=FALSE)
# store output
assign(paste("cr", i, sep=""), cr)
}The code above reruns the CR algorithm on the dataset with 70, 60, 50 … and 10 occurrences as the quota of the subsampling. Then the results can be plotted with:
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB richness", ylim=c(0,90))
for(i in quotas){
current <- get(paste("cr", i, sep=""))
lines(stages$mid[1:94], current $divCSIB, col="red")
}
As the subsampling quota decreases, so does the mean value of richness, and also the relative deviation of the series. The Shareholder Quorum subsampling approach (Alroy, 2010) effectively circumvents this problem by using a different proxy for sampling completeness, other than the number of occurrences. Rather than looking at the basic unary information of occurrences, the SQS method assesses sampling with frequency coverage. Coverage is an intuitive way to express the completeness of sampling. In the species sampling pool all species have a frequency (Fk) that sum up to 1. In this sense, the coverage of a sample is the sum of species frequencies in the original sampling pool (Fk), but only for those species that actually have been sampled. This rule effectively coerces sampling coverage between 0 and 1. Different levels of sampling can be created by maximizing the desired coverage of a sample. This desired coverage is referred to as the ‘quorum’. After the work of Chao and Jost (2012), ecologists usually refer to this approach as coverage-based rarefaction.
5.4.3.1. The ‘inexact’ approach to SQS
The original description of the SQS algorithm ensured different levels of coverages by going through the occurrences (or collections) randomly and aggregating the estimated frequencies of the species that are taken from the sample until this sum reached the subsampling quorum. John Alroy (2014) referred to this approach as the ‘inexact’ method to perform SQS. This is the default way to do SQS.
# sqs with 0.6 quorum
sqs0.6 <-subsample(fossGen, iter=50, q=0.6,
tax="genus", bin="stg", type="sqs")
#sqs with 0.3 quorum
sqs0.3 <-subsample(fossGen, iter=50, q=0.3,
tax="genus", bin="stg", type="sqs")
# plotting
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB richness", ylim=c(0,175))
lines(stages$mid[1:94], sqs0.6$divCSIB, col="blue")
lines(stages$mid[1:94], sqs0.3$divCSIB, col="green")
lines(stages$mid[1:94], dd$divCSIB, col="black")
legend("topleft", legend=c("raw", "SQS, q=0.6", "SQS, q=0.3"),
col=c("black", "blue", "green"), lwd=c(2,2,2), bg="white")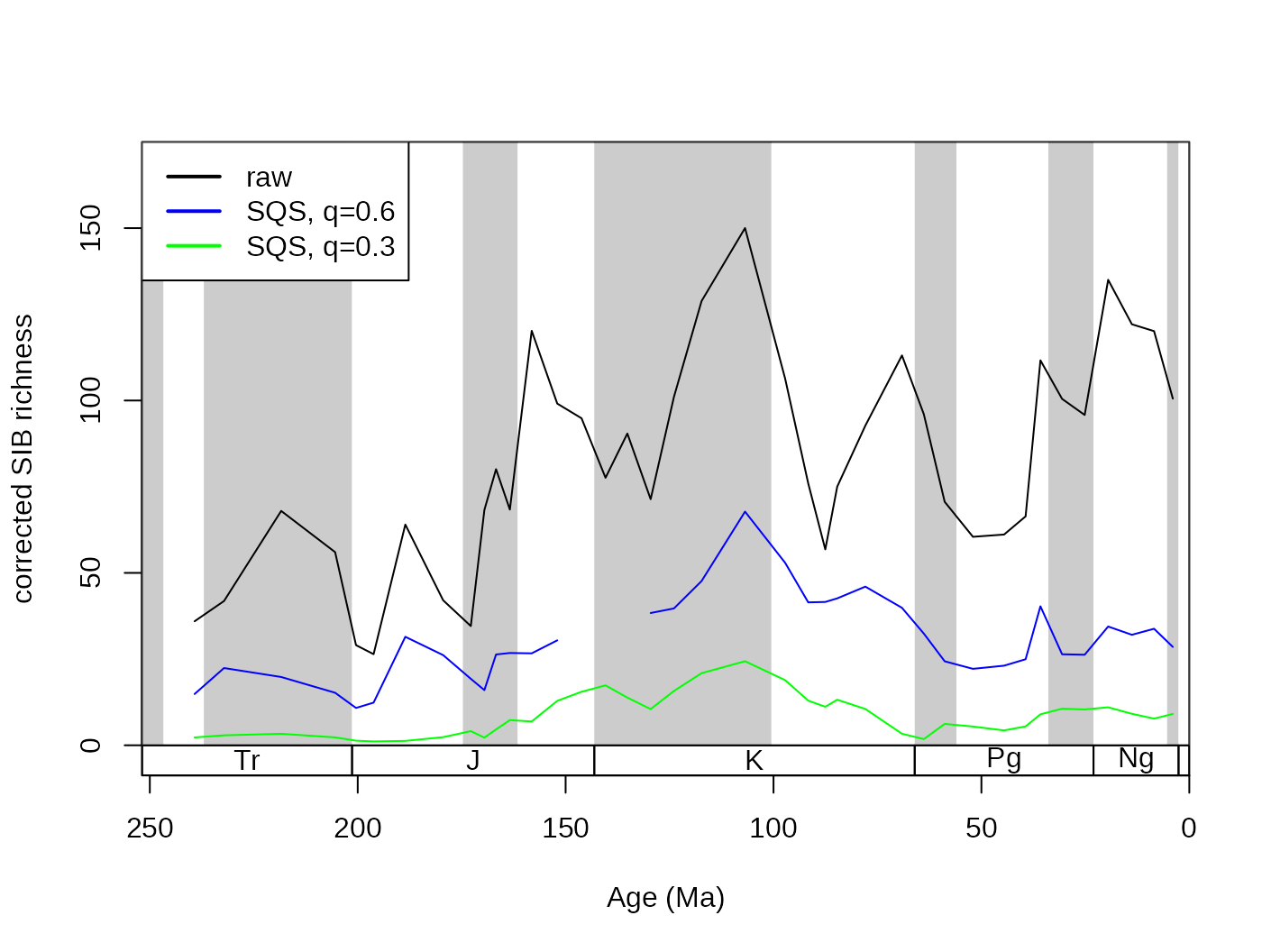 This procedure is dependent on the proper estimation of the species’
frequencies in the sampling pool (?frequencies). Alroy (2010) suggested
that the observed frequencies should be adjusted with Good’s total
sample coverage estimator u, which is dependent on the number
of occurrences (o) and the number of single-collection
(single-occurrence) taxa (1O ).
This procedure is dependent on the proper estimation of the species’
frequencies in the sampling pool (?frequencies). Alroy (2010) suggested
that the observed frequencies should be adjusted with Good’s total
sample coverage estimator u, which is dependent on the number
of occurrences (o) and the number of single-collection
(single-occurrence) taxa (1O ).
\[u = 1 - {1}O/o\]
This is the first correction of Alroy (2010, for overall coverage).
He also suggested that for Paleobiology Database occurrences, a
different version of this estimator (u’) might be more accurate
that is estimated using the single reference taxa, instead of
single-collection taxa. This estimator is then used to estimate the true
frequencies of species. You can choose between these versions by
toggling the singleton argument between "occ"
(default), "ref" and it can be switched off by setting it
to FALSE. Note that "ref" will require you to
provide a reference variable (ref argument). The
discrepancy between the methods is very small:
sqsCollSing <-subsample(fossGen, iter=50, q=0.4,
tax="genus", bin="stg", type="sqs",
singleton="occ")
sqsRefSing <-subsample(fossGen, iter=50, q=0.4,
tax="genus", bin="stg", type="sqs", ref="reference_no",
singleton="ref")
sqsNoSing <-subsample(fossGen, iter=50, q=0.4,
tax="genus", bin="stg", type="sqs", singleton=FALSE)
# plotting
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB richness", ylim=c(0,100))
lines(stages$mid[1:94], sqsRefSing$divCSIB, col="blue")
lines(stages$mid[1:94], sqsCollSing$divCSIB, col="black")
lines(stages$mid[1:94], sqsNoSing$divCSIB, col="red")
legend("topleft", legend=c("SQS, q=0.4", "SQS, q=0.4, ref", "SQS, q=0.4, occ"),
col=c("black", "blue", "red"), lwd=c(2,2,2), bg="white")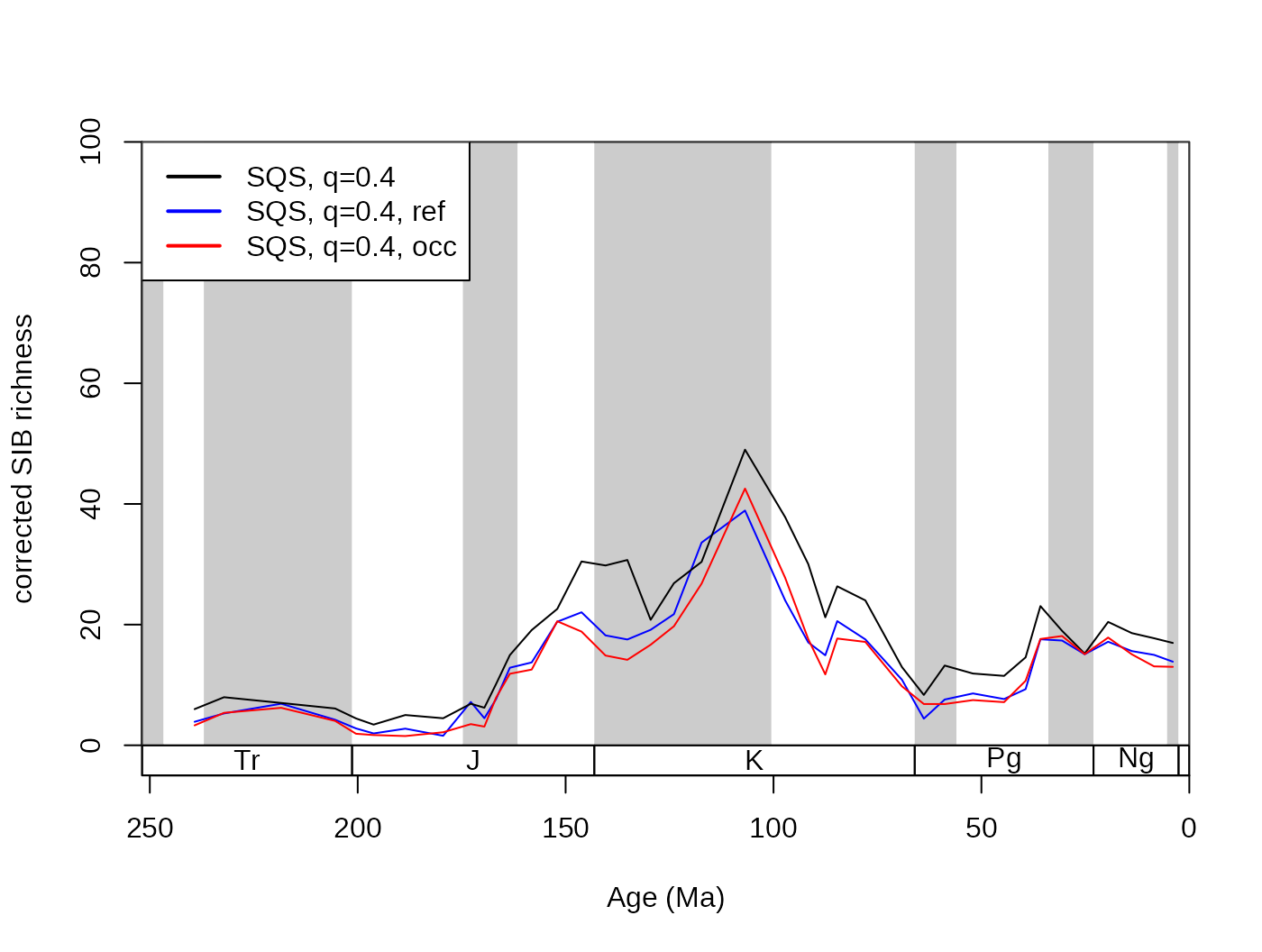 Alroy (2010) also mentioned two additional corrections (for Evenness
and Dominance and Single large collections) that also have
minor effects on the overall results. You can use the
Alroy (2010) also mentioned two additional corrections (for Evenness
and Dominance and Single large collections) that also have
minor effects on the overall results. You can use the
excludeDominant and largestColl arguments to
turn these on. Note that you have to provide a collection variable to
make largestColl work, which is only available if
excludeDominant=TRUE.
sqsPure <-subsample(fossGen, iter=50, q=0.5,
tax="genus", bin="stg", type="sqs")
sqsCorr <-subsample(fossGen, iter=50, q=0.5,
tax="genus", bin="stg", ref="reference_no",coll="collection_no",
type="sqs", singleton="ref", excludeDominant=T, largestColl=T)
# plotting
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB richness", ylim=c(0,100))
lines(stages$mid[1:94], sqsPure$divCSIB, col="blue")
lines(stages$mid[1:94], sqsCorr$divCSIB, col="black")
legend("topleft", legend=c("uncorrected SQS",
"corrected SQS"), col=c("blue", "black"),
lwd=c(2,2), bg="white")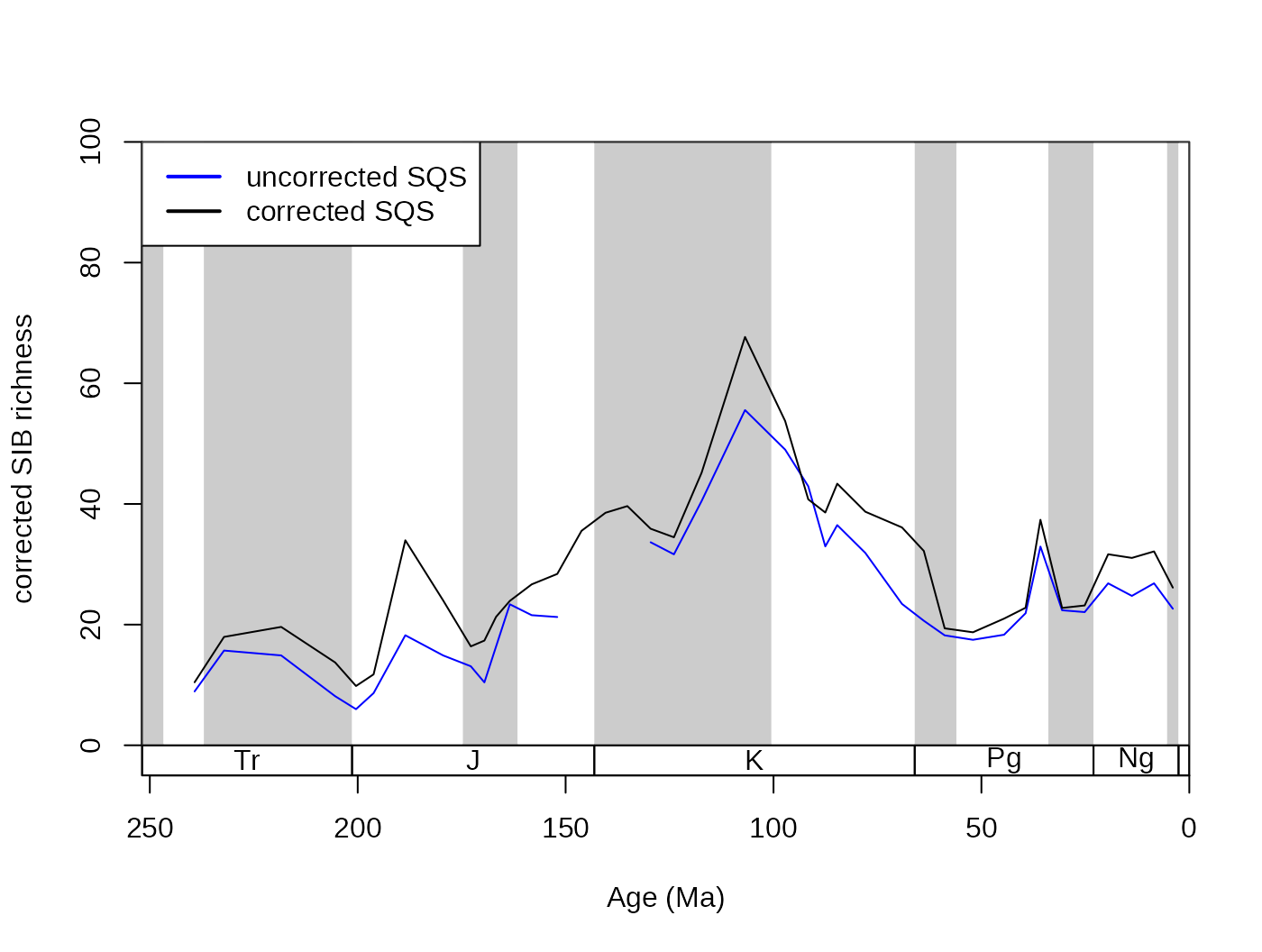
So far, these calculations were based on occurrence-based
rarefaction, but SQS can be calculated similarly to OW, by tallying the
occurrences collection-by-collection. This can be enforced with the
byList=TRUE option.
sqsByColl <-subsample(fossGen, iter=50, q=0.5,
tax="genus", bin="stg", ref="reference_no",coll="collection_no",
type="sqs", singleton="ref", excludeDominant=T, largestColl=T, byList=TRUE)
# plotting
tsplot(stages, shading="series", boxes="sys", xlim=52:95,
ylab="corrected SIB richness", ylim=c(0,100))
lines(stages$mid[1:94], sqsByColl$divCSIB, col="blue")
lines(stages$mid[1:94], sqsCorr$divCSIB, col="black")
legend("topleft", legend=c("by-list SQS",
"by-occurrence SQS"), col=c("blue", "black"),
lwd=c(2,2), bg="white")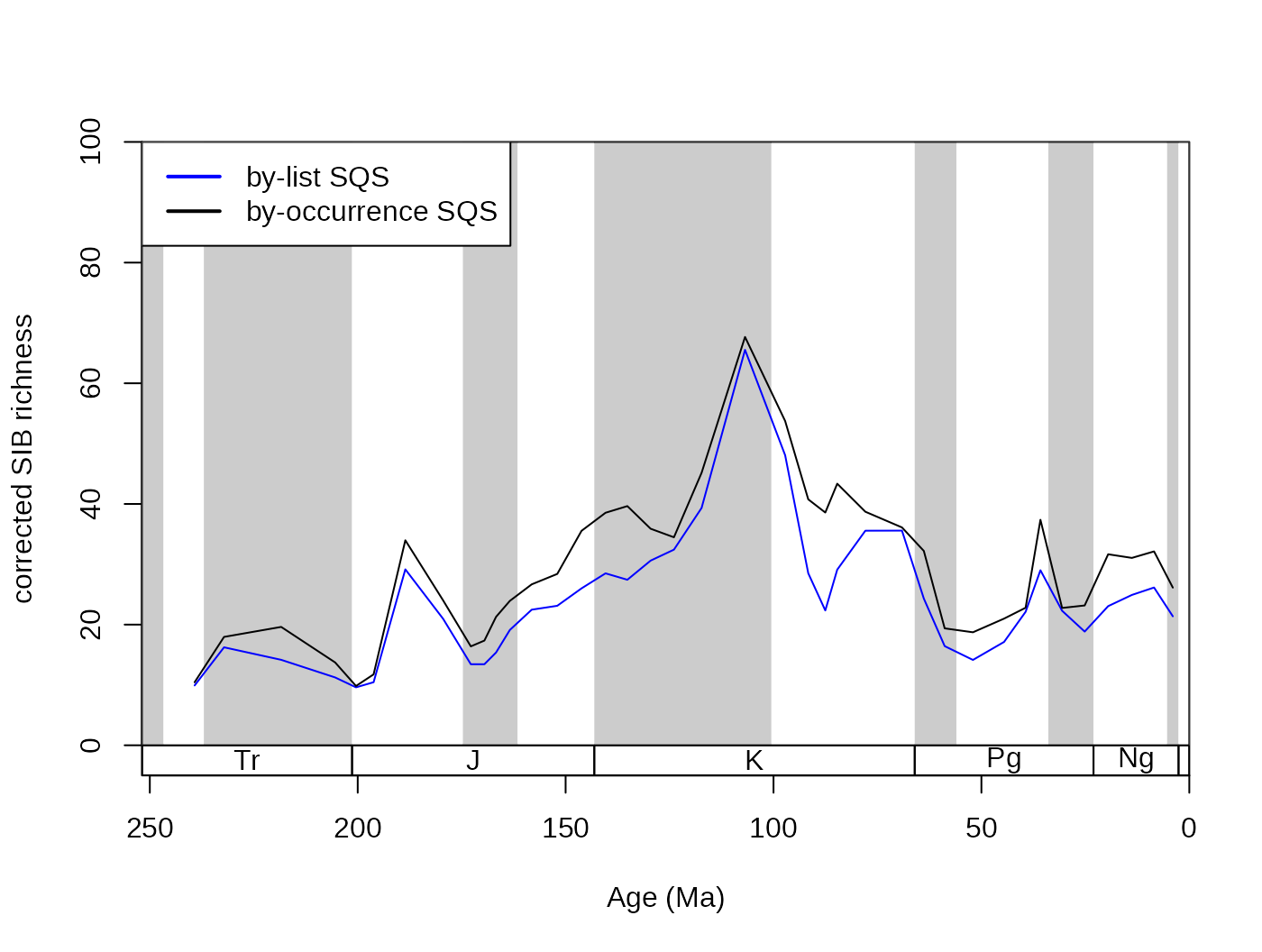
5.4.3.2. Concluding remarks on SQS
SQS is probably the most appropriate method we have today for
sampling standardization of diversity curves. Although the number of
potential influencing factors is high, they do not influence the overall
trajectory of the curves, and the induced variation is almost equal to
the variability that is created by subsampling itself (plot some results
with output="dist"). The systematical evaluation of these
options is still lacking, but we are working on the appropriate testing
framework.
5.4.4. Subsampling trial functions
The different subsampling types can be contrasted by comparing the
subsampling trial functions (subtrialCR(),
subtrialOXW() and subtrialSQS()).The
procedures are of these functions are also implemented within the
subsample() function, and they can be used to produce one
‘trial dataset’ from the total sampled data. The output of these
function is a logical vector indicating the rows of the data that should
be present in one particular trial dataset. Future versions of the
package will allow the writing and application of custom subsampling
trial functions, with which the subsample() function will
be entirely customizable.
6. Taxon-specific characteristics
6.1. Environmental affinities
Most taxa prefer certain environments. Our understanding of
environmental affinities of different taxa depend on the characteristics
of sampling. In some questions the definition of two contrasting
environments suffices, and most hypotheses can be tested with univariate
statistics between two samples. Using the most basic logic, one could
infer that a taxon preferred environment A to environment
B, all other things being equal, if it occurs more in that
particular environment over its lifetime. This is sometimes referred to
as the majority rule of affinity, and is very straightforward to
implement. In our test example, a number of variables are added based on
the raw data, which express the environmental conditions of the dataset.
For the sake of demonstration, the bathymetric affinities
bath will be calculated. First, we have to omit those
occurrences that have unknown depth conditions.
knownBath <- fossils[fossils$bath!="uk",]6.1.1. Majority rule
After this simple filtering, the affinity() function can
be applied to the dataset, with specifying the usual arguments, plus the
"majority" method. The function also needs to be notified
about the column (its name. env="bath" in our case) that
contains the binary environmental variable in question:
affMajor <- affinity(knownBath, bin="stg", tax="genus",
method="majority", coll="collection_no", env="bath")
table(affMajor)## affMajor
## deep shal
## 25 693The output of this function is a single character vector, values
specifying the apparently preferred environments and the
names attribute defining the taxon names. NA
entries occur when the function is unable to define an affinity
(i.e. the number of occurrences from both environments is the same). It
is not mandatory to provide collection identifiers (coll).
If these are given, then the multiple occurrences of a taxon within a
collection (sample) will be treated as one. As you can see, an
overwhelming majority of the taxa have shallow affinities based on this
method, but this is not surprising, as most of the occurrences come from
shallow environments.
table(knownBath$bath)##
## deep shal
## 1035 256446.1.2. Binomial method
If sampling is so dominated by shallow occurrences, then the obvious
question that arises is whether the apparent pattern of affinity only is
present because this biased sampling. In order to correct for this
‘background’ sampling issue, Foote (2006) suggested that the environment
affinity should be determined by comparing the taxon’s observed pattern
of occurrences to that present in the total dataset (null sampling
probability of the environment). This approach was also used by
Kiessling and Aberhan (2007) and Kiessling and Kocsis (2015) in later
studies. The sampling probability of the environment is calculated by
taking the proportion of occurrences in the two environments in the
total dataset from the range of the taxon. Under random sampling
conditions, the number of total occurrences and the number among these
that come from an environment is modelled by a binomial distribution.
Given a certain level of confidence one can guess whether the sampled
occurrences of a taxon represent a significantly higher or lower
proportion than predicted by the null probability. The more likely it is
that a simple binomial model can produce the observed success/total
trial ratio, the less we know about the affinity of the taxon. This
approach is implemented by the default "binom" method.
affBin1 <- affinity(knownBath, bin="stg", tax="genus",
method="binom", coll="collection_no", env="bath")
table(affBin1)## affBin1
## deep shal
## 135 574The significance of the binomial tests can be chosen by the
alpha argument that toggles the breadth of proportions that
is considered to be output by the randomness of sampling. The default
alpha = 1 setting (above) will not perform the binomial
test. It will only compare the taxon’s observed proportion of
occurrences from the different environments to those observed in the
total dataset. The lower this alpha value is, the less taxa
will get an assigned affinity.
affBin0.5 <- affinity(knownBath, bin="stg", tax="genus",
method="binom", coll="collection_no", env="bath", alpha=0.5)
table(affBin0.5)## affBin0.5
## deep shal
## 127 149
affBin0.1 <- affinity(knownBath, bin="stg", tax="genus",
method="binom", coll="collection_no", env="bath", alpha=0.1)
table(affBin0.1)## affBin0.1
## deep shal
## 77 56Using the standard 95% percent as alpha level is an arbitrary choice.
As the fossil record has quality issues, a 90% confidence
(alpha = 0.1) is preferred in most cases. It is also
possible to do the calculations based on the collection counts, instead
of the number of occurrences. For these the argument bycoll
has to be set to TRUE.
6.2. Changing characteristics
The entire occurrence dataset can be subdivided into time-bin and taxon-specific subsets. We can use each of these subsets to describe the taxa’s ecological characteristics if we assert that these occurrences are contemporaneous.
6.2.1. Geographic range
One of the most ecologically relevant characteristics of a taxon is
its total geographic range, which can be calculated with the
georanges() function, using a number of methods. These rely
on the geographic coordinates of the occurrences, which are in this case
reconstructed internally by the Paleobiology Database (with GPlates).
This function is applicable to time-slice-specific subset of a single
taxon, such as the Pleistocene occurrences of the genus
Acropora
oneTax <- corals[corals$stg==94 & corals$genus=="Acropora",]
georange(oneTax, lat="paleolat", lng="paleolng", method="co")## co
## 213This function call outputs coordinate occupancy, the number of unique
pairs of coordinates the taxon has. Besides this very simple one,
multiple additional methods are available by calling functions from the
icosa and vegan packages. You can inspect
these with ?georange.
6.2.2. The tabinate() iterator function
For large-scale analyses of taxa, it is desirable to apply the
function above to all time-bin/taxon specific subsets of the data. This
can be desired for numerous calculations that use contemporaneous
occurrences of a taxon as input, which can be iterated for every
time-bin/taxon subset with the tabinate() function. To use
this function you have to provide the taxon and time bin identifiers
(tax and bin, respectively), and the function
(FUN) that needs to be iterated on every taxon/time-bin
subset of the dataset. This function has to take x as the
primary argument (the database subset), but additional arguments will be
passed to it.
For instance, if you are interested in the number of collections each
taxon has in each time slice, instead of using a combination of
functions (subset(), unique() and
table()) you can define a function, which can be iterated
on all subsets.
collCount <- function(x) length(unique(x$collection_no))
allGeo <- tabinate(corals, bin="stg", tax="genus", FUN=collCount)This is a somewhat slower solution, but its utility is evident when
more complicated functions are passed to the iterator, such the
georanges() presented before.
allGeo <- tabinate(corals, bin="stg", tax="genus",
FUN=georange, lat="paleolat", lng="paleolng", method="co")The tabinate() function currently accepts functions that
output single values or vectors as output, but its applicability will be
increased in the future.
Acknowledgements
The development of the package began in the context of a project on global paleobiogeography funded by the Deutsche Forschungsgemeinschaft (DFG project numbers Ko 5382/1-1, Ko 5382/1-2, Ki 806/16-1). The package is also part of the DFG Research Unit 2332: TERSANE (TEmperature Related Stressors in ANcient Extinctions). We are thankful to all testers of the package including Emilia Jarochowska, Vanessa Roden, Andreas Lauchstedt, Gwen Antell and Roger Benson for suggestions, and all participants of the first year International Master’s Programme for Paleobiology at the GeoZentrum Nordbayern, FAU in Erlangen, Germany. We also thank all enterers of the Paleobiology Database.
References
Alroy, J. (2008). Dynamics of origination and extinction in the marine fossil record. Proceedings of the National Academy of Science, 105, 11536–11542.
Alroy, J., Marshall, C. R., Bambach, R. K., Bezusko, K., Foote, M., Fürsich, F. T., … Webber, A. (2001). Effects of sampling standardization on estimates of Phanerozoic marine diversification. Proceedings of the National Academy of Science, 98(11), 6261–6266. https://doi.org/10.1073/pnas.111144698
Alroy, J., Aberhan, M., Bottjer, D. J., Foote, M., Fürsich, F. T., Harries, P. J., … Visaggi, C. C. (2008). Phanerozoic Trends in the Global Diversity of Marine Invertebrates. Science, 321(5885), 97–100. https://doi.org/10.1126/science.1156963
Alroy, J. (2010). The Shifting Balance of Diversity Among Major Marine Animal Groups. Science, 329, 1191–1194. https://doi.org/10.1126/science.1189910
Alroy, J. (2010b). Geographical, environmental and intrinsic biotic controls on Phanerozoic marine diversification. Palaeontology 53:1211-1235.https://doi.org/10.1111/j.1475-4983.2010.01011.x
Alroy, J. (2014). Accurate and precise estimates of origination and extinction rates. Paleobiology, 40(3), 374–397. https://doi.org/10.1666/13036
Alroy, J. (2015). A more precise speciation and extinction rate estimator. Paleobiology, 41(04), 633–639. https://doi.org/10.1017/pab.2015.26
Bell, M. A. (2015). geoscale: Geological Time Scale Plotting. R package version 2.0. Retrieved from https://CRAN.R-project.org/package=geoscale
Foote, M., & Raup, D. M. (1996). Fossil Preservation and the Stratigraphic Ranges of Taxa. Paleobiology, 22(2), 121–140.
Chao, A., & Jost, L. (2012). Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology, 93(12), 2533–2547. https://doi.org/10.1890/11-1952.1
Foote, M. (1999). Morphological Diversity In The Evolutionary Radiation Of Paleozoic and Post-Paleozoic Crinoids. Paleobiology, 25(S2), 1–115.
Foote, M. (2000). Origination and Extinction Components of Taxonomic Diversity: General Problems. Paleobiology, 26(4), 74–102.
Foote, M. (2006). Substrate Affinity and Diversity Dynamics of Paleozoic Marine Animals. Paleobiology, 32(3), 345–366. https://doi.org/10.1666/05062.1
Good, I. J. (1953). The Popoulation Frequencies of Species and the Estimation of Population Parameters. Biometrika, 40(3/4), 237–264.
Gradstein, F. M., Ogg, J. G., & Schmitz, M. D. (2020). The geologic time scale 2020. Elsevier.
Kiessling, W., & Aberhan, M. (2007). Environmental determinants of marine benthic biodiversity dynamics through Triassic-Jurassic time. Paleobiology, 33(3), 414–434.
Kiessling, W., & Kocsis, A. T. (2015). Biodiversity dynamics and environmental occupancy of fossil azooxanthellate and zooxanthellate scleractinian corals. Paleobiology, 41(3), 402–414.
Kiessling, W., & Simpson, C. (2011). On the potential for ocean acidification to be a general cause of ancient reef crises. Global Change Biology, 17, 56–67. https://doi.org/10.1111/j.1365-2486.2010.02204.x
Kocsis, A. T., Reddin, C. J., Alroy, J. and Kiessling, W. (2019). The R package divDyn for quantifying diversity dynamics using fossil sampling data. Methods in Ecology and Evolution. https://doi.org/10.1111/2041-210X.13161
Raup, D. M. (1975). Taxonomic Diversity Estimation Using Rarefaction. Paleobiology, 1, 333–342. https://doi.org/10.1017/S0094837300002633
Raup, D. M. (1979). Biases in the fossil record of species and genera. Bulletin of the Carnegie Museum of Natural History, 13, 85-91.
Raup, D. M. (1985). Mathematical Models of Cladogenesis. Paleobiology, 11(1), 42–52.
Sepkoski Jr, J. J. (2002). A compendium of fossil marine animal genera. Bulletins of American Paleontology, 363, 1-560.